Ключевые звенья патогенеза остеопороза
Кость – сложная динамическая система, в которой постоянно и одновременно протекают процессы резорбции и формирования костной ткани. Состояние скелета в целом, определяющееся его начальным морфогенезом, а также ремоделированием костной ткани на протяжении жизни, зависит от скоординированной регуляции и активности формирующих кость клеток – остеобластов (ОБ) и резорбирующих клеток – остеокластов (ОК).
Остеобластогенез
Остеобласты представляют собой мононуклеарные клетки, которые вырабатывают костный матрикс (остеоид), состоящий преимущественно из коллагена I типа, и обеспечивают его минерализацию. Остеобласты образуются из незрелых остеопрогениторных клеток в надкостнице и костном мозге, которые вырабатывают основной регуляторный фактор транскрипции RUNX2 (фактор транскрипции 2, содержащий домен Runt, также известен как CBF-a1). Дифференцировка остеопрогениторных клеток происходит под влиянием множества факторов роста, включая костные морфогенетические белки (BMP), факторы роста фибробластов (FGF; преимущественно FGF18), тромбоцитарный фактор роста (PDGF) и трансформирующий фактор роста β (TGF-ß).
После дифференцировки остеобласты вырабатывают остеогенные маркеры, включая фактор транскрипции Sp7 (ранее известный как остерикс), Col1A1, костный сиалопротеин 2 (BSPII), макрофагальный колониестимулирующий фактор 1 (M-CSF), костную щелочную фосфатазу (ALP), остеокальцин (также известный как костный протеин Gla), остеопонтин (OPN), RUNX2, медиаторы сигнального пути Wnt/ß-катенин, N-терминальный пропептид коллагена I типа и SPARC (также известный как остеонектин).
Образование зрелыми остеобластами костной ткани стимулируется инсулиноподобным фактором роста II (IGF-II) и TGF-ß.
Остеобласты, окруженные костным матриксом, превращаются в остеоциты, после чего они прекращают синтез остеоида и минерализацию костного матрикса; вместо этого такие клетки обеспечивают паракринную регуляцию активных остеобластов, а также, по всей видимости, угнетают образование остеокластов и резорбцию костной ткани.
Остеокластогенез
Остеокласты, которые образуются из клеток моноцитарно-макрофагального ряда, интенсивно вырабатывают тартрат-резистентную кислую фосфатазу типа 5 (TRAP) и катепсин K.
Окончательно дифференцированные зрелые остеокласты фенотипически характеризуются экспрессией специфических маркеров, таких как TRAP, и рецепторов к кальцитонину. В участках резорбции костной ткани клеточная мембрана остеокластов образует складки («гофрированная мембрана»), которые увеличивают площадь резорбции. Фермент TRAP, который секретируется гофрированной мембраной, вызывает дефосфорилирование остеопонтина (OPN), а также стимулирует миграцию остеокластов и резорбцию костной ткани. Ионы кальция и фосфата, которые высвобождаются при разрушении гидроксиапатита (основного компонента минерализованного матрикса), собираются в небольшие везикулы и выделяются во внеклеточную жидкость. Маркеры резорбции костной ткани включают сывороточный C-телопептид коллагена I типа и мочевой N-телопептид коллагена I типа.
В регуляции активности остеокластов участвуют: гормоны, включая паратиреоидный гормон (ПТГ), кальцитонин и ИЛ-6; растворимые факторы, такие как M-CSF (дефицит этого фактора вызывает остеопетроз); факторы транскрипции, такие как c-Fos, NFATcl и NFkB; лиганд протеинового рецептора, активирующего ядерный фактор kB (RANKL; также известен как член 11 суперсемейства TNF (TNFSF11)). Процесс костной резорбции включает также синтез цистеиновых протеиназ, таких как катепсин K и матриксные металлопротеиназы (MMP). MMP-9 и MMP-14 стимулируют миграцию остеокластов к костной поверхности. Дефицит эстрогена усиливает костную резорбцию, тогда как недостаточное поступление и низкая плазменная концентрация витамина K, а также недостаточное карбоксилирование остеокальцина сопровождаются снижением минеральной плотности костной ткани (МПКТ) и увеличением риска переломов.
Патология
Нарушение указанной регуляции может привести к тяжелым нарушениям состояния скелета, характеризующимся как снижением (например, остеопороз), так и повышением (например, остеопетроз) массы кости. Количество активных остеокластов определяется дифференцировкой и слиянием прекурсоров этих клеток, а также их гибелью за счет апоптоза. Повышение пула активных остеокластов, сопровождающееся повышением костной резорбции и снижением массы кости, наблюдается при остеопорозе.
Эти процессы реализуются путем активации многогранных межклеточных событий, включающих регуляцию посредством транскрипционных нуклеарных факторов (семейство NF kappa) генов, кодирующих медиаторы, способные регулировать метаболизм, образование и резорбцию кости, и их рецепторы (Ota N., Hunt S.C., Nakajima T. et al., 2000). Наиболее значимым эффектом медиаторов является их действие как локальных аутокринных и паракринных факторов, что обеспечивается совпадением условий стимуляции секреции цитокинов и усиления экспрессии рецепторов на клетках-мишенях (Mundy G.R., 2000).
Инициальное событие в костном ремоделировании – увеличение остеокластной костной резорбции, связанной с повышенной клеточной адгезией между остеокластными предшественниками и костномозговыми стромальными клетками или остеобластами (Tanaka Y., Maruo A., Fujii K. et al., 2000).
Главный клеточный процесс в патологической костной резорбции – остеокластная активность. Костная резорбция – уникальная функция остеокластов.
Остеокласты – специфические макрофагальные полинуклеары. Отвечая на интегрин-опосредованные сигналы, остеокласты образовывают специализированную формацию, устанавливающую ограниченное микроокружение между ними и костью, где и происходит матричная деградация путем процесса, включающего протоновый транспорт.
Цитокины являются истинными медиаторами ремоделирования костной ткани (Jergensen N.R., 1997).
Центральную роль среди многих факторов (см. табл. 5), стимулирующих чрезмерную остеокластную активность, играют цитокины, такие, как интерлейкин-1 (ИЛ-1), фактор некроза опухоли α (ФНО-α), обладающие провоспалительными свойствами.
Таблица 5
Системные, локальные и другие факторы, регулирующие костное ремоделирование

Цитокины типа ИЛ-6 также стимулируют остеокластное образование путем активации гликопротеиновой рецепторной субъединицы gp130 на стромальных⁄остеобластных клетках, которая ведет к активации экспрессии рецепторного активатора лиганда нуклеарного фактора kappa β.
Все локальные факторы, за исключением простагландина, имеют белковую природу и синтезированы различными типами клеток, имеющими мезенхимное происхождение.
Открыты новые представители семейства лигандов ФНО-рецепторов, а именно: рецепторный активатор нуклеарного фактора kappa β (RANK) и RANK-лиганд (RANKL). Их взаимодействия важны для дифференцировки остеокластов из гемопоэтических предшественников в физиологических и патологических условиях (Roux S., Orcel P., 2000).
В окончательный путь остеокластогенеза было предложено включать три составляющие системы цитокинов: активатор рецепторов ядерного фактора Кβ лиганд (RANKL), его рецептора – активатора ядерного фактора Кβ (RANK) и его растворимого рецептора-приманки, остеопротегерин (ОПГ).
RANKL является цитокиноподобной молекулой, активирующей мембраносвязанные рецепторы на поверхности преостеокластов.
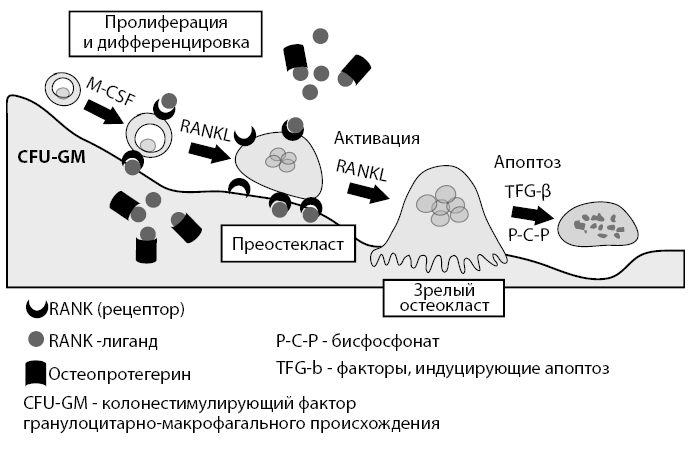
Рисунок 9. Схема созревания остеокласта и взаимодействие RANK-лиганд с рецепторами и остеопротегерином
RANK-лиганд активируя рецепторный активатор нуклеарного фактора kappa β, приводит к стимуляции созревания остеокластов.
В то же время иной пептид – остеопротегерин – является «рецептором-ловушкой», который связывает RANK-лиганд и предупреждает таким образом активирующее влияние последнего на RANK.
Остеопротегерин имеет очень мощное тормозящее действие на образование остеокластов.
Именно исходя из таких представлений, данный механизм регуляции резорбирующей активности остеокластов рассматривают как новую перспективную возможность для лечения заболеваний, сопровождающихся снижением костной массы и активной резорбцией, а также повышением риска переломов (Г.Я. Шварц, 2004).
Патогенетические взаимосвязи кардиоваскулярных заболеваний и остеопороза
По данным зарубежных публикаций, у пациентов, перенесших остеопоретический перелом шейки бедра, в 70 % имется патология сердечно-сосудистой системы (H. Diamond, S. Thornley, R. P. Smerdely, 1997). В нашей клинике таких пациентов было более 80 %, при этом ни один из них не знал о своем заболевании ранее и соответственно лечение и профилактику остеопороза не проводил (Верткин А.Л., Наумов А.В., 2006). Аналогичные данные приводит и И.В. Буданова и соавт. (2005).
По мнению Насонова Е.Л. (1999), сердечно-сосудистые заболевания (ССЗ), дегенеративные заболевания позвоночника и суставов, а также остеопороз, являющиеся наиболее частыми проявлениями инволютивной патологии, следует определять как «кальцийдефицитные» болезни человека. Это объясняется определенным сходством патогенеза остеопороза и атеросклероза, при которых поврежденные моноцитарные клетки, в одном случае дифференцируются в сосудистой стенке в макрофагоподобные «пенистые» клетки, в другом – в остеокласты.
Помимо этого, костная и сосудистая ткани имеют ряд и других общих морфологических и молекулярных свойств. Сосудистый кальцификат представлен теми же элементами, что и костная ткань: соли кальция, фосфаты, связанные с гидроксиапатитом, остеопонтин, костный морфогенный белок, матриксный Gla-белок, коллаген типа I, остеонектин, остеокальцин и др. (Shanahan C.M., Cary N.R., Metcalfe J.C. et al., 1994; Giachelli C.M., Bae N., Almeida M. et al., 1993; Bostrom К., Watson K.E., Hom S. et al., 1993; Katsuda S., Okada Y., Minamoto T. et al., 1992). Более того, стенка артерии, пораженной атеросклерозом, состоит из предшественников остеобластов, которые обладают способностью синтезировать минеральные компоненты, характерные для костной ткани (Parhami F., Morrow A.D., Balucan J. et al, 1997).
Особенно большое значение имеет тот факт, что окисленные липопротеины низкой плотности (ЛПНП), принимающие участие в развитии атеросклеротического поражения сосудов (Witztum J.L., Steinberg D., 1991), стимулируют минерализацию, опосредованную как костными остеобластами, так и остеобластоподобными клетками, изолированными из сосудистой стенки.
В исследованиях Christoph R. et al (2000), в популяции 91 611 пациентов, методом случай – контроль было продемонстрировано снижение риска переломов у пациентов, получающих статины (0,55; 95 % ДИ, 0,44–0,69), при том, что у пациентов, принимавших фибраты, снижение риска переломов не наблюдалось.
В исследованиях Uyama O., Hansen M.A. было показано, что у пациентов с низкой минеральной плотностью кости (МПК) чаще наблюдается повышение уровня липидов, приводящее к более тяжелому коронарному атеросклерозу и высокому риску инсульта и инфаркта миокарда (Uyama O., Yoshimoto Y., Yamamoto Y., Kawai A. 1997; Р. van der Recke P., Hansen M.A., Hassager C., 1999). У женщин в ранний период постменопаузы (P. van der Recke и et al., 1999) снижение МПКТ на одно стандартное отклонение от пиковой костной массы (0,4 г/см2) ассоциируется с увеличением риска общей летальности на 43 % (относительный риск – ОР=1,4; pр<0,05) и преждевременной смерти от сердечно-сосудистой патологии в течение 17 лет наблюдения (ОР=2,3; pр<0,05).
В эпидемиологическом исследовании, проведенном Browner W.S., Sooley D.G., Vogt T.M. (1991), в которое были включены 9704 женщины старше 65 лет, каждое снижение МПКТ проксимального участка лучевой кости на одно стандартное отклонение от нормы увеличивало риск преждевременной смерти от инсульта в течение последующих 2 лет (не связанный с остеопоретическими переломами) на 40 % (Browner W.S., Pressman A.R., Nevitt M.C. et al., 1993).
В исследовании G.N. Farhat, A.B. Newman, K. Sutton-Tyrrell et all. (the Health ABC study, 2007) у пациентов c кардиоваскулярными заболеваниями выявлены более низкие показатели МПК в телах позвонков, шейки бедра и дистальном отделе предплечья, особенно у пациентов c высоким уровнем провосполительных цитокинов (Vasan R.S., Sullivan L.M., Roubenoff R., Dinarello C.A., Harris T., Benjamin E.J., Sawyer D.B., Levy D., Wilson P.W., D’Agostini R.B. (2003). Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction; Framingham Heart Study. Circulation 107:1486–1491).
В исследовании U. Sennerby, B. Farahmand, A. Ahlbom, S. Ljunghall, K. Michaëlsson (Osteoporos Int., feb. 2007) было выявлено 2–3-кратное увеличение риска перелома проксимального отдела бедренной кости у пациентов c ССЗ в анамнезе.
Следует подчеркнуть, что ряд авторов относят потерю МПК к категории предикторов ССЗ, а именно поражений коронарных артерий (Marcovitz P.A., Tran H.H., Franklin B.A., O’Neill W.W., Yerkey M., Boura J., Kleerkoper M., Dickinson C.Z. (2005). Usefulness of bone mineral density to predict significant coronary artery disease. Am J Cardiol 96:1059–1063).
В ряде исследований отмечена общность патогенеза артериальной гипертонии и остеопороза. В частности, активность ренин – ангиотензиновой системы (РАС), с одной стороны, за счет влияния на локальный кровоток и кровоснабжение костей, вызывает вазоконстрикцию микроциркуляторного русла, а с другой – оказывает непосредственное влияние на выработку ангиотензина ІІ. Последний является фактором роста, непосредственно стимулирующим пролиферацию остеокластов и увеличивающим уровень эндотелина-1, содержание которого при активации РАС увеличивается не только в эндотелии, но и в остеокластах (Sasaki T., Hong M.H., 1993). Эти данные подтверждаются в клинике остеопротективном действии ингибиторов АПФ (Stimpel M., Jee W.S.S., Yamamoto N. et al., 1995). Эти препараты, подавляя активность ангиотензина ІІ, способствуют меньшей резорбции остеокластов костной ткани, снижая ее потери МПКТ (Hatton R., Stimpel M., Chambers T.J., 1997; Stimpel M., Hatton R., Chambers T.J., 1997).
Все эти данные позволяют предположить, что нарастание частоты остеопороза, эктопической кальцификации и атеросклероза в целом у одних и тех же пациентов невозможно объяснить только неспецифическими возрастными факторами, обусловливающими независимое накопление этих патологических состояний в пожилом возрасте. Вероятно, они еще имеют и общую патогенетическую основу (Pahmani F., Garfinkel A., Demer L.L., 2000).
Кальцификация сосудов является независимым фактором риска сердечно-сосудистых заболеваний (ССЗ).
Кальцификация любой артерии или клапана сердца повышает риск сердечно-сосудистых событий и смертности в 3–4 раза, а также считается прогностическим фактором ишемической болезни сердца (ИБС). Отложение кальция в коронарных артериях может привести к ослаблению вазомоторных реакций и нарушить стабильность атеросклеротических бляшек. У пациентов с нестабильной стенокардией либо инфарктом миокарда чаще наблюдается множество небольших очагов отложений кальция, тогда как у пациентов со стабильной стенокардией – несколько крупных очагов.
Кальцификация коронарных артерий легко определяется при помощи КТ; оценка кальцификации может использоваться в клинических исследованиях для прогнозирования риска сердечно-сосудистых событий.
Аналогично кальцификация аорты также является независимым прогностическим фактором сердечно-сосудистых событий.
Кальцификация стенок артерий сопровождается снижением их эластичности, что приводит к значительной заболеваемости и смертности от таких заболеваний, как артериальная гипертензия, аортальный стеноз, гипертрофия миокарда, инфаркт миокарда и ишемия нижних конечностей.
В настоящем обзоре рассматриваются патогенетические механизмы, общие для остеопороза и кальцификации сосудов, а также обсуждаются клинические данные в отношении частоты развития кальцификации сосудов и риска сердечно-сосудистых событий у пациентов с остеопорозом.
Выделяют четыре типа кальцификации сосудов – интимы, средней оболочки (медии) и клапанов сердца, а также кальцифицирующую артериолопатию.
Наиболее распространенной формой является кальцификация интимы, которая развивается под воздействием проатерогенных и провоспалительных факторов, таких как липопротеины и цитокины.
Кальцификация медии, которая ассоциируется с возрастом, сахарным диабетом и хронической болезнью почек (ХБП), приводит к увеличению риска ампутации и смертности от сердечно-сосудистых причин по сравнению с пациентами, у которых кальцификация сосудов отсутствует.
При кальцификации медии эластин распадается с образованием метаболитов, которые активируют формирование клетками отложений кальция, тогда как неповрежденный эластиновый матрикс стабилизирует фенотип гладкомышечных клеток (ГМК) сосудистой стенки in vivo.
Кальцификация митрального кольца, которая является наиболее распространенной формой кальцификации клапанов сердца (на втором месте – аортальный клапан), положительно коррелирует с атеросклерозом и смертностью вследствие сердечно-сосудистых причин независимо от тяжести ИБС.
Кальцифицирующая артериолопатия является тяжелой и жизнеугрожающей формой кальцификации сосудов, которая, как правило, возникает у пациентов с гиперпаратиреоидизмом или на поздних стадиях ХБП, сопровождаясь развитием панникулита и некроза кожи. При этом смертность достигает 80 %, что обусловлено прогрессирующей ишемией кожи и сепсисом. В редких случаях возможно также поражение брыжейки и ткани легких.
Факторы риска поражения эндотелия, такие как дислипидемия, артериальная гипертензия, сахарный диабет или воспалительные цитокины, образующиеся в избыточной жировой ткани, усиливают экспрессию веществ, стимулирующих адгезию лейкоцитов к внутренней поверхности стенки артерии и их соединение с эндотелиальными и ГМК. Основным результатом такой воспалительной реакции является миграция ГМК из медии в интиму, их пролиферация и трансформация в остеобластоподобные клетки с развитием кальцификации и атеросклероза.
В этом процессе также участвуют моноциты и дендритные клетки, которые вырабатывают MMP в ответ на различные продукты окисления и воспалительных реакций.
При изучении кальцинированных сосудов человека были обнаружены маркеры остеогенеза (ALP, остеокальцин, BSPII, коллаген II), факторы транскрипции остеобластов (RUNX2, Sp7, MSX2) и фактор транскрипции хондроцитов SOX9. Инактивация гена RUNX2 приводила к выраженному уменьшению кальцификации ГМК сосудов.
Кальцификация атеросклеротических бляшек интимы происходит по механизму энхондральной оссификации; процесс кальцификации медии более сходен с эндесмальной оссификацией, поскольку: а) изначально минерализация происходит в матриксных везикулах, связанных с волокнами внеклеточного матрикса; б) отсутствует необходимость предшествующего наличия хрящевой ткани; в) основную роль в процессе минерализации играет BMP-2.
Кальцификация аортального клапана ассоциируется с фенотипом остеобластов, поскольку при кальцификации в клапанах определяются повышенные концентрации мРНК, кодирующей OPN, BSPII, остеокальцин и RUNX2. Культивирование интерстициальных клеток аортального клапана человека в стимулирующей остеогенез питательной среде, содержащей BMP-2, BMP-4, BMP-7 и TGF-ß, в течение 21 дня привело к остеобластной дифференцировке интерстициальных клеток клапана, что определялось на основании повышенной активности и экспрессии ALP.
BMP участвуют в дифференцировке остеобластов, одновременно оказывая влияние на синтез АФК и усиливая адгезию моноцитов к сосудистой стенке. Действие этих белков блокируется MGP, витамин K-зависимым белком, который также угнетает минерализацию сосудистой стенки в качестве кофактора α2-HS-гликопротеина (также известен как фетуин-A). RANKL является ключевым фактором созревания остеокластов, а также препятствует кальцификации. OPG предотвращает связывание RANKL с его рецептором.
Сигнальный путь Wnt, который играет важную роль в дифференцировке остеобластов, угнетается склеростином и DKK-1. В сосудистой стенке Wnt активируется фактором транскрипции MSX2, который нейтрализует ингибирующий эффект DKK-1, что приводит к усилению кальцификации сосудов.

Рисунок 10 (адаптирован по C.E. Lampropoulos, I. Papaioannou, D.P. D’Cruz, 2012). Общие патогенетические механизмы остеопороза и кальцификации сосудов
Ионы фосфата, которые проникают через стенку ГМК вместе с котранспортером Pit-1, непосредственно стимулируют кальцификацию сосудов, тогда как ионы пирофосфата служат ингибитором кальцификации. OPN связывает ионы кальция и гидроксиапатита, ингибируя формирование кристаллов и кальцификацию сосудов; этот белок взаимодействует с интегриновыми рецепторами, что приводит к активации остеокластов. ПТГ угнетает активацию остеобластов и усиливает резорбцию костной ткани; посредством активации PKA этот гормон стимулирует дифференцировку остеобластов и минерализацию клеток сосудистой стенки.
Витамин D увеличивает поступление кальция в клетки сосудистой стенки, что приводит к кальцификации. Окисленный холестерин ЛПНП стимулирует экспрессию мощных медиаторов дифференцировки остеокластов. Ангиотензин II участвует в активации остеокластов. Сокращения: BMP = костный морфогенетический белок; Ca2+ = кальций; DKK-1 = белок Dickkopf-1; E-NPP1 = экзонуклеотид-пирофосфатаза (член 1 семейства фосфодиэстераз); M-CSF = макрофагальный колониестимулирующий фактор 1; MGP = матриксный Gla-белок; OPG = остеопротегерин; OPN = остеопонтин; Pi = неорганический фосфат; PPi = пирофосфат; PKA = протеинкиназа A; ПТГ = паратиреоидный гормон; PTH1R = рецептор к паратиреоидному гормону типа 1; RANKL = лиганд рецептора, активирующего ядерный фактор kB; АФК = активные формы кислорода; RUNX2 = транскрипционный фактор 2 с доменом runt; ГМК = гладкомышечная клетка; TRAP = тартрат-резистентная кислая фосфатаза типа 5.
Перициты мелких кровеносных сосудов, а также интерстициальные клетки аортального клапана человека могут дифференцироваться в остеобласты, хондроциты и адипоциты, что способствует возникновению и прогрессированию кальцификации сосудов. Минерал, который откладывается в кальцинированных бляшках сосудистой стенки, представляет собой гидроксиапатит. Низкое поступление витамина K (который является необходимым кофактором для активации некарбоксилированного матриксного Gla-белка (MGP)) и высокая концентрация некарбоксилированного MGP тесно связаны с повышенным риском кальцификации сердечно-сосудистой системы и смертности.
Полученные к настоящему времени данные поддерживают гипотезу о наличии связанных метаболических каскадов для процессов остеопороза и кальцификации сосудов. Эти два состояния, по всей видимости, развиваются постепенно и необратимо прогрессируют с возрастом.
Таблица 6
Клинические доказательства связи между кальцификацией сосудов и риском переломов


Таблица 7
Клинические доказательства связи остеопороза и сердечно-сосудистых событий



Таблица 8
Клинические доказательства связи остеопороза и кальцификации сосудов


Сокращения: МПК – минеральная плотность костной ткани.
Данная связь может объясняться тем, что кальцификация сосудов, которая приводит к снижению кровотока либо к ограничению физической активности, сопровождается нарушением костного метаболизма, что, в свою очередь, ведет к потере костной массы.
В исследованиях на крысах было продемонстрировано, что бисфосфонаты в дозах, сравнимых с угнетающими костную резорбцию, подавляют кальцификацию артерий и клапанов сердца, не оказывая влияния на сывороточные концентрации кальция и фосфатов.
Этот эффект объясняется защитным действием бисфосфонатов в отношении сосудистой стенки, включая повышение чувствительности макрофагов к факторам, вызывающим апоптоз, и предупреждение образования пенных клеток посредством угнетения захвата холестерина ЛПНП.
В опытах на крысах с артериальной гипертензией бисфосфонаты тормозили развитие атеросклероза и пролиферацию ГМК сосудов. В проспективном клиническом исследовании бисфосфонаты замедляли прогрессирование атеросклеротических бляшек и увеличение индекса кальцификации брюшного отдела аорты (AAC) у женщин с остеопорозом, в то время как у здоровых женщин без лечения наблюдалось прогрессирование кальцификации сосудов; это указывает на защитный эффект бисфосфонатов в отношении атеросклероза.
В исследовании кальцификации брюшного отдела аорты MESA было показано, что, после поправки на возраст и факторы риска, более низкая МПК ассоциировалась с повышенным индексом кальцификации коронарных артерий у женщин, а также у пациентов обоих полов с повышенным значением индекса AAC.
В другом исследовании, в котором выполнялось изучение коронарных артерий методом мультиспиральной компьютерной томографии, была установлена связь индекса кальцификации коронарных артерий и выраженности атеросклеротических бляшек с низкой МПК у женщин в пре– и постменопаузе; данная связь не зависела от возраста и сердечно-сосудистых факторов риска. В исследовании продолжительностью 9 лет, которое включало 236 женщин, потеря костной массы в период менопаузы была значимо выше у женщин с прогрессированием кальцификации аорты по сравнению с отсутствием прогрессирования.
У пациентов с почечной недостаточностью, получавших гемодиализ, была установлена статистически значимая отрицательная корреляция между уровнем ремоделирования кости, который определялся в образцах, полученных методом костной биопсии, и индексом кальцификации коронарных артерий.
По-видимому, у пожилых женщин низкая МПК шейки бедренной кости является маркером выраженных атеросклеротических изменений.
Во многих исследованиях была показана связь между остеопорозом и толщиной интимы-медии сонной артерии, которая является хорошо известным фактором риска сердечно-сосудистых заболеваний.
У женщин с остеопорозом наблюдалось нарушение функции эндотелия плечевой артерии по сравнению со здоровыми женщинами, что определялось на основании поток-зависимой вазодилатации после реактивной гиперемии.
Ригидность стенки артерии, которая измерялась на основании плече-лодыжечной скорости распространения пульсовой волны, ассоциировалась с остеопорозом и атеросклерозом коронарных артерий, который определялся при помощи мультиспиральной КТ.
Кроме того, в ряде клинических исследований была продемонстрирована связь между кальцификацией сосудов и риском остеопоротического перелома (таблица 3), а также между остеопорозом и сердечно-сосудистыми событиями (таблица 4).
В исследовании MINOS с участием 781 мужчины в возрасте ≥ 50 лет, в котором период наблюдения составлял 10 лет, более высокие значения AAC ассоциировалось с возрастанием риска переломов в 2–3 раза, независимо от величины МПК либо наличия падений в анамнезе.
В группе, включавшей 2348 здоровых женщин в постменопаузе, связь между выраженностью кальцификации аорты (которая определялась при помощи КТ) и потерей костной массы была статистически значимой и не зависела от возраста; кроме того, у женщин с наличием кальцификации вероятность перелома позвонков возрастала в 5 раз и перелома шейки бедра – в 3 раза по сравнению с женщинами, у которых кальцификация отсутствовала.
В популяционном когортном исследовании, которое включало 2662 здоровые женщины в постменопаузе, за период наблюдения продолжительностью 7,5 года выраженная кальцификация аорты ассоциировалась со снижением МПК и увеличением риска перелома проксимальной части бедренной кости в 2,3 раза.
Увеличение риска переломов, особенно позвонков, и скорость снижения МПК также положительно ассоциировались с прогрессированием кальцификации аорты.
У женщин с остеопорозом риск инсульта в 4,8 раза выше по сравнению с женщинами с нормальной МПК.
В другом исследовании на основании когорты Framingham у женщин со сниженной МПК наблюдалось более выраженное увеличение AAC на протяжении 25-летнего периода наблюдения. У женщин с выраженной ИБС (определяемой как сужение просвета крупного сосуда > 50 %) остеопороз, по-видимому, служил независимым прогностическим фактором данного заболевания.
В исследовании с участием 2576 женщин в постменопаузе ускоренная потеря костной массы шейки бедренной кости ассоциировалась с увеличением риска смертности от сердечно-сосудистых причин.
Была установлена независимая корреляция между МПК, повышенным риском переломов и поражением периферических артерий.
В популяционном когортном исследовании, включавшем 16 294 пациента, было показано, что сердечная недостаточность ассоциируется с факторами, которые связаны с ускоренной потерей костной массы и повышением риска переломов, в частности перелома шейки бедра (в 4 раза); этот результат не должен вызывать удивления, поскольку в большинстве случаев пациенты с сердечной недостаточностью характеризуются ограничением подвижности.
Общие патогенетические механизмы
Факторы, участвующие в патогенезе как остеопороза, так и кальцификации сосудов, включают белки, гормоны, химические элементы, липиды и витамины (табл.).
В таблице 9 суммированы общие для обоих процессов механизмы, которые обсуждаются далее.
Таблица 9
Общие патогенетические факторы для остеопороза и кальцификации сосудов


Сокращения: BMP – костный морфогенетический белок; MGP – матриксный Gla-белок; ОПГ – остеопротегерин; ОПН – остеопонтин; ПТГ – паратиреоидный гормон; RANKL – лиганд рецептора, активирующего ядерный фактор kB.
Костные морфогенетические белки (BMP)
BMP, представители суперсемейства TGF-ß, индуцируют дифференцировку мезенхимальных клеток по остеобластной линии, что сопровождается усилением синтеза коллагена. Кроме того, эти белки подавляют экспрессию коллагеназы-3 остеобластами, что приводит к уменьшению распада коллагена и сохранению костной массы.
BMP-2 вызывает дифференцировку остеобластов посредством индукции фактора транскрипции MSX2. BMP-6 опосредует стимулирующие эффекты глюкокортикоидов в отношении дифференцировки остеобластных клеток, поскольку лечение глюкокортикоидами приводит к значительному повышению концентрации мРНК BMP-6 и экспрессии данного белка.
В процессе формирования костной ткани BMP-2 и BMP-7 индуцируют экспрессию RUNX2 и Sp7; кроме того, эти белки стимулируют транскрипцию белка ноггина, который, обладая высоким сродством к BMP, связывается с ними и нейтрализует их биологические эффекты; по-видимому, этот механизм ауторегуляции ограничивает активность BMP в остеобластах.
BMP оказывают провоспалительное и прооксидантное действие в системных артериях. В исследованиях было подтверждено значительное повышение активности BMP в очагах атеросклеротических поражений. BMP-2 вырабатывается клетками сосудистого эндотелия и ГМК под влиянием провоспалительных факторов, таких как TNF и пероксид водорода.
В регуляции экспрессии BMP-2 центральную роль играет сигнальный путь NF-kB. Кроме того, активация NF-kB в эндотелии и увеличение экспрессии BMP-2 и TNF были продемонстрированы при гипергомоцистеинемии.
BMP-2 вызывает эндотелиальную дисфункцию и стимулирует выработку в эндотелиальных клетках большого количества активных форм кислорода (АФК) под действием НАДФН-оксидазы, что приводит к активации эндотелия и к усилению адгезии моноцитов.
В опытах на модели сахарного диабета у мышей было показано, что стимуляция BMP-2 and MSX2 сопровождается усилением кальцификации сосудов, а диета с высоким содержанием жиров стимулирует экспрессию MSX1 и MSX2 в периваскулярных адвентициальных клетках. BMP-4, содержание которого гораздо выше в легочных артериях по сравнению с сосудами большого круга, вызывает выраженную эндотелиальную дисфункцию системных артерий с явлениями вазоконстрикции, артериальной гипертензии и развитием атеросклеротических бляшек, в то время как легочные артерии остаются интактными. Была установлена связь стимуляции BMP-4 с развитием атеросклероза и артериальной гипертензии, тогда как прерывание сигнального пути BMP-4 ассоциировалось с развитием легочной гипертензии. Антагонисты BMP (включая фоллистатин, ноггин и MGP), которые вырабатываются в эндотелиальных клетках периферических артерий, регулируют активность BMP в сосудистой стенке. У мышей с ХБП введение BMP-7 сопровождалось значительным уменьшением кальцификации аорты и снижением гиперфосфатемии. Тем не менее размеры очагов атеросклеротических поражений не уменьшались. Введение BMP-7 приводило к снижению экспрессии остерикса.
Сигнальный путь RANKL-RANK-OPG
RANKL вырабатывается стромальными клетками и остеобластами и является ключевым фактором дифференцировки моноцитарно-макрофагальных предшественников остеокластов в многоядерные остеокласты, а также активации зрелых остеокластов.
RANKL активирует антиапоптозную серин-треониновую киназу Akt (также известную как протеинкиназа B) посредством сигнального комплекса, включающего Src-киназу и ассоциированный с рецептором TNF фактор 6 (TRAF6). Связывание RANKL с его рецептором на клетках-предшественниках остеокластов приводит к активации NFkB и NFATc1, которые необходимы для дифференцировки остеокластов. Активация NFkB происходит практически сразу, а NFATc1 – через 24–48 ч после связывания RANKL с рецептором. RANKL приводит к образованию АФК, включая ионы кислорода, свободные радикалы и пероксиды – как неорганические, так и органические, которые играют крайне важную роль в процессе остеокластогенеза. RANKL индуцирует также выработку каспазы-3 – фермента, вовлеченного в процесс апоптоза; при угнетении активности каспазы-3 остеокласты теряют способность к дифференцировке в ответ на воздействие RANKL.