Глава I Патофизиологическая, биохимическая и клиническая характеристики хронической алкогольной интоксикации
Негативные эффекты этилового алкоголя обусловлены нарушениями обмена веществ в клетках различных органов, которые возникают вследствие постоянного повреждающего действия как самого яда и его метаболитов (ацетоальдегида, уксусной кислоты и алкогольных эфиров жирных кислот), так и продуктов других патологических продуктов, возникающих при действии этилового алкоголя (свободных радикалов, агрессивных молекул средней молекулярной массы и т. д.). Рассматривая злоупотребление алкоголем в прикладном аспекте, целесообразно выделить четыре компонента в повреждающем действии этих веществ:
– воздействие на внеклеточный матрикс,
– мембранный,
– лиганд-рецепторный,
– метаболический.
Подобное разделение позволяет раскрыть суть патохимических реакций, которые лежат в основе клинических проявлений хронической интоксикации этиловым алкоголем, а также объяснить новые подходы в их диагностике и лечении.
Взаимодействие с элементами внеклеточного матрикса
Всасываясь и распределяясь в организме человека, этиловый алкоголь изменяет биохимические процессы, протекающие в микроокружении клеток, в частности во внеклеточном матриксе (ВКМ). Компоненты последнего секретируются самими клетками и выполнены коллагенами, гликопротеинами и гелем, образованным водой и гидратированными в ней небольшими по размерам белковыми молекулами. В матриксе присутствуют биологически активные вещества, в том числе цитокины. Скопление биологически активных молекул в микроокружении клеток представляет собой организованную систему, которая играет роль биорегулятора, по крайней мере в пределах одногруппных кластеров клеток, и обеспечивает протекание биохимических реакций в клеточных мембранах и в самих клетках. Более того, передача биохимической информации через ВКМ имеет черты сходства с лиганд-рецепторным взаимодействием, подобным тому, которое происходит в синаптических и гормональных системах, только участники взаимодействия представлены элементами матрикса, плазматической мембраной и цитоскелетом клетки. Таким образом, образуется самостоятельная регуляторная система со своей структурно-функциональной единицей, которая наряду с другими системами организма осуществляет регуляцию его гомеостаза, в частности такие фундаментальные процессы, как деление клеток, их морфогенез, пролиферацию и т. д. (Ямсков И. А., 2001). Эта система является одной из первых мишеней для действия этилового алкоголя.
Подробности взаимодействия этанола с компонентами ВКМ мало изучены. Однако ясно то, что, по крайней мере, этиловый алкоголь может растворяться во внеклеточной воде, нарушать ее коллоидные и проводниковые свойства и приводить к изменению состава водных секторов. Молекулы этанола конкурируют с водой за связывание с различными составляющими ВКМ. В основе этой конкуренции лежит способность этанола и воды к образованию водородных связей, однако этанол является амфифильным соединением и, в отличие от воды, способен одновременно связываться с гидрофильными и гидрофобными центрами матрикса. Это приводит к вытеснению воды из внеклеточного сектора, к изменению конформационного строения молекул ВКМ и к нарушению состава супрамолекулярной организации и функции в целом. Известно также, что алкоголь нарушает активность матричных металлопротеиназ и ослабляет адгезию некоторых специфических субстратов ВКМ (Partridge, 1999).
Приведенные рассуждения имеют отношение к нескольким клиническим проблемам. Назовем две из них: особенности проведения инфузионной терапии при алкогольной патологии, а также получение и применение принципиально новых лекарственных веществ, выделенных из компонентов ВКМ, которые предназначены для лечения алкогольного абстинентного синдрома (ААС) и хронического алкоголизма в целом. Точкой приложения здесь являются гликопротеины ВКМ (например, нейролин, гепалон и др.).
Взаимодействие с липидами плазматических мембран Свободно проникая к самым различным клеткам, этанол оказывает действие на их плазматические мембраны. Он легко внедряется в их липидный бислой, связывается с конгломератами липидов мембран, «разделяет» жирнокислотные цепи фосфолипидов и увеличивает внутримембранные пространства бислоя. Одним из наиболее важных клинических последствий этого процесса является обезвоживание мембраны, которое происходит за счет вытеснения молекул воды (в среднем до двух из двадцати молекул), формирующих нормальную гидратную оболочку каждого из мембранных липидов (Ulrich et al., 1994). Подчеркнем, что в наибольшей степени вытесняются те молекулы воды, которые образуют важные водородные связи с карбоксильными группами соседних липидов и во многом определяют структурные, физико-химические свойства последних и биологических мембран в целом.

Рис. 1. Взаимодействие молекул этанола с фосфатидилхолином (молекулярное моделирование по П. П. Якуцени, 2002)
Условные обозначения: Показано типичное расположение фосфолипида и окружающих его пяти молекул этанола. Молекулы № 1, 2 и 3 образуют водородные связи (светло-серый цвет) с карбоксильными группами жирнокислотных остатков (темно-серый цвет). Отражены три из восьми возможных мест связывания этанола или воды. Молекулы этанола № 4, 5 образуют водородные связи с фосфатной частью полярной головки липидов (темный цвет). Отражены два из более чем десяти возможных мест связывания. Кислород выделен темным оттенком. Вертикальная (алкильная) цепочка липида (атомы водорода этой молекулы не показаны) уходит за нижнюю границу рисунка.
Известно, что мультислои фосфатидилхолина даже при нулевой относительной влажности содержат одну молекулу воды на один липид. При 15 % относительной влажности связываются две молекулы воды с липидом (Ивков В. Г., 1981), из них: 1,2 ± 0,3 молекулы воды взаимодействуют с фосфатной группой липида, а 0,7 ± 0,2 молекулы – с карбоксильными группами его жирнокислотных остатков. Считается, что межмолекулярные водородные связи образуются вторыми из указанных молекул воды. Выступая в роли своеобразных мостиков, они связывают карбоксильные группы соседних липидов.
При помощи молекулярного моделирования, отражающего взаимодействие одиночной молекулы фосфолипида (в данном случае – фосфатидилхолина) с молекулами этанола, П. П. Якуцени (2002) показал, что этанол также способен связываться с фосфатными группами фосфолипидов и карбоксильными группами жирнокислотных цепей (см. рис. 1). Расчеты, выполненные с помощью методов молекулярной фармакологии и квантовой механики, позволили предположить, что молекулы этанола способны вытеснять молекулы «связанной» воды из фосфолипидов биологических мембран и тем самым нарушать естественную структуру. Однако, в отличие от воды, этанол располагает только одной гидроксильной (OH) группой и не способен образовать мостика, связывающего молекулы соседних липидов друг с другом. Рабочая гипотеза П. П. Якуцени (2002) о механизме дегидратирующего действия этилового алкоголя, первично связанного со структурами липидного бислоя, подтверждена более поздними углубленными расчетами (см. рис. 2).

Рис. 2. Характерное распределение этанола в липидной мембране при концентрации алкоголя в плазме крови на уровне 1‰ (результат молекулярного моделирования П. П. Якуцени, 2002)
Условные обозначения:
Показана часть липидов верхней половины бислоя. Тяжелые атомы обозначены серым цветом, атомы водорода – светло-серым, кислород – темным цветом. Верхние молекулы этанола (№ 1–4) находятся в зоне преимущественного связывания, ниже которого расположен интерфазный слой мембран, содержащий протоноакцепторы. Нижняя молекула этанола (молекула № 5) вызывает дефект упорядоченности алкильных «хвостов» липидных молекул.
Оказалось, что образование водородных связей в наибольшей степени происходит на уровне интерфазного слоя липидной мембраны, в котором имеется большое количество функциональных групп – протоноакцепторов (эфирных и карбоксильных), способных фиксировать ионы водорода, т. е. там, где расположены функционально важные полярные головки фосфолипидов (см. верхнюю часть рис. 2). В мембранах непьющих людей между этими головками существует «пояс» межмолекулярных связей, образованный молекулами водорода и воды, который обеспечивает каркас липидных головок. Постоянное присутствие этанола в плазме крови на уровне 1‰, в первую очередь, разрушает этот молекулярный «пояс» и является центральным эффектом этанола, разупорядочивающего структуру липидной мембраны. Увеличение концентрации этанола в плазме крови до 2 % и более вытесняет оставшиеся молекулы связанной с липидами воды и изменяет их гидратную оболочку. Это приводит к нарушению композиции биологических мембран клеток. Длительная алкоголизация приводит к насыщению мембран спиртом и к изменению конформаций жирнокислотных цепочек липидов, что сопровождается структурными дефектами гидрофобной части мембран клеток.
Представленный процесс является элементом физико-химического механизма адаптации организма человека, возникающей при хронической интоксикации этиловым алкоголем.
В контексте рассматриваемой проблемы нарушение гидратной оболочки липидной части мембран клеток приводит к двум клиническим последствиям:
– сдвигу водных секторов между вне– и внутриклеточным пространством и развитию дисгидрии, порой жизнеопасной;
– изменению чувствительности рецепторов к лекарственным препаратам.
Специфику дегидратации при ААС необходимо учитывать при проведении инфузионной терапии, а изменение фармакодинамики лекарственных веществ – в проведении медикаментозной терапии таких больных.
В мембранах клеток рецепторы и ферментные комплексы фиксированы липидами, как якорями. Если белки не закреплены в мембране, то они «плавают» в ее липидном бислое, как льдинки в воде. В результате постоянного присутствия этанола некоторые из них «тонут» и становятся неспособными взаимодействовать со своими эндогенными лигандами (или лекарственными препаратами).
В мембранотоксическом действии этанола есть еще одна особенность, затрагивающая деятельность клеток всех типов. Липидный слой плазматических мембран представлен гликолипидами, холестерином и фосфолипидами. Последних – большинство, и они играют наиболее важную роль в обмене веществ в нейронах, глие, гепатоцитах, миокардиоцитах и других клетках. В состав фосфолипидов входят фосфатидилхолин (лецитин), фосфатидилэтаноламин, фосфатидилинозит, сфигномиелин и другие вещества, которые при ферментативном распаде образуют ряд важнейших биологически активных соединений или их предшественников. Например, из фосфатидилинозита образуются два вторичных посредника, инозитол-3-фосфат и диацилглицерин, необходимые для передачи медиаторных, гормональных и других информационных сигналов клеткам. Фосфатидилхолин (лецитин) служит предшественником синтеза холина, ацетилированная форма которого (ацетилхолин – медиатор) осуществляет нейротрансмиссию в нервной системе и защищает ее от избыточных адренергических воздействий. Это свойство лецитина интенсивно изучается с целью поиска способов фармакологической защиты печени, мозга и сердца от повреждений, возникающих при алкоголизации.
Удельный вес компонентов липидного бислоя в семействах различных клеток варьируется. Изменения, возникающие в них под действием этилового спирта, развиваются в соответствии с их биохимическим составом. Попадая в кровь, продукты деградации липидов могут индуцировать некоторые ферменты (например, усиливать активность фосфотидилэтаноламинметилтрансферазы) и служить источниками образования эндоперекисей, пагубная роль которых при алкогольной болезни в последние годы убедительно доказана.
Чем длительнее прием алкоголя, тем боульшие повреждения в мембранах он вызывает, вплоть до полного исчезновения определенных субстратов в некоторых из них. Например, сравнительный морфологический анализ липидного состава биомембран, проведенный на препаратах большого мозга и мозжечка неалкоголиков и алкоголиков, показал отсутствие у последних плазмалогенов в плазматических мембранах головного мозга (Leasch, 1972). (Плаз-малогены – липидные соединения, входящие в состав миелина глиальных клеток и обеспечивающие изолирующие свойства их отростков.) Возможно, поэтому проведение инфузионной терапии больным с абстинентным синдромом натрийсодержащими растворами может сопровождаться активацией демиелинирующих процессов в нервной ткани и приводить к инвалидизации таких больных (Illowsky et al., 1987).
В результате нарушаются матричная и транспортная функции мембран клеток, страдает трансмембранный перенос веществ в клетки и координация деятельности последних в целом.
Липиды мембран являются липофильными веществами, поэтому в водной среде они агрегируют за счет гидрофильных воздействий и Ван-дер-Ваальсовых сил. Жирорастворимость этанола резко снижается по мере его разбавления водой, вплоть до 50 % при 40 %-ной концентрации. Сопоставление этих двух факторов позволяет сделать вывод о том, что чем крепче спиртные напитки, тем большее мембраноповреждающее действие они оказывают. Особенно это касается напитков более крепких, чем водка.
Взаимодействие с рецепторами биологических мембран различных клеток
Помимо поражения плазматических мембран клеток, этиловый алкоголь оказывает действие на их рецепторы. Последние представлены интегральными белками, иногда очень сложными по своей структуре. Они регулируют прохождение ионов и биологически активных веществ через мембраны нейронов и клеток исполнительных органов (так называемые ионотропные рецепторы) и участвуют в обмене веществ в клетках, осуществляя эволюционно закрепленные каскадные биохимические реакции, которые начинаются в мембранах клеток (так называемые метаболотропные рецепторы). В организме человека рецепторы синтезируются, транспортируются к своим «местам узнавания» – в мембраны клеток и органелл. Этот процесс контролируется определенными генами, следовательно, плотность белков-рецепторов в пределах своих локусов закреплена наследственно и является различной у разных людей. С этим обстоятельством связывают предрасположенность людей к алкоголизму, поскольку определенные медиаторные системы головного мозга и их рецепторы участвуют в нейроадаптивном сдвиге, возникающем в организме человека при частом употреблении им спиртных напитков.
Действие алкоголя приводит к нейрохимическим изменениям в пределах локусов мозга с наибольшей плотностью тропных к нему рецепторов, таких как НМДА-, дофамин-, опиатергических, серотониновых и др. Однако последствия возбуждения рецепторов могут быть компенсированы еще пока не нарушенными метаболическими реакциями, протекающими в цитоплазме и ядрах клеток (например, глюконеогенез, гликолиз, синтез НАД, эвакуация ионов водорода, нейтрализация эндоперекисей и др.). Каждая группа рецепторов координирует определенные функции в организме человека. Постоянное возбуждение рецепторных систем может приводить к нарушению этих функций. Некоторые их них представлены на рисунке 3.
Продолжающаяся алкоголизация вызывает изменения, превышающие «систему надежности» организма, что влечет за собой развитие самых различных нарушений.
Изучение механизмов формирования алкогольной мотивации является одной из наиболее важных проблем в нейрохимии и нейробиологии алкоголизма, сложность решения которой заключается в разнообразии методических подходов для ее решения и трудностях в интерпретации психобиологических, психофизиологических и нейрохимических взаимосвязей, возникающих при такой патологии.

Рис. 3. Взаимодействие этанола с некоторыми рецепторами
Разнонаправленные исследования, выполненные в этой области знаний, указывают на то, что в формировании мотивации к употреблению алкоголя и появлении аддиктивного поведения, характеризующегося непреодолимой тягой человека к приему спиртного, главная роль принадлежит медиаторным системам головного мозга. Подчеркнем, что подавляющее большинство лекарственных веществ, применяемых в современной терапии алкоголизма, направлено на восстановление нейрогенной адаптации или нормоергии между многообразием возбуждающих и тормозных структур ЦНС.
В процессе развития зависимости от этанола в наибольшей степе ни изучена роль опиоидных, катехоламин-, серотонин-, ГАМК-, НМДАергических систем головного мозга человека. Уже на ранних стадиях ее формирования возникает разлад в сбалансированной работе возбуждающих и тормозных медиаторных систем. Возрастает активность НМДА-рецепторов в определенных структурах ЦНС (преимущественно в миндалине, гиппокампе, n. accumbens), увеличивается выход дофамина и норадреналина из соответствующих нейронов, а высвобождение ацетилхолина, серотонина и ГАМК в определенных участках мозга снижается.
Каждому из перечисленных медиаторов свойственно контролировать определенную совокупность процессов, протекающих в головном мозге и в периферических функциональных системах организма человека. Сбой равновесия на уровне нейрональных цепей изменяет взаимосвязи медиаторов с другими аутокоидами ЦНС, что влияет на состояние самого головного мозга и других систем организма, таких как сердечно-сосудистая, эндокринная, дыхательная и т. д.
Изменение активности медиаторных систем сопровождается нарушением динамики трансмембранных ионных потоков и кинетики биохимических реакций в клеточном окружении и в самих клетках (с изменением ионного состава которых связывают многие побочные эффекты лекарственных веществ, применяемых в период детоксикации). Некоторые ионы выполняют функцию коферментов в метаболических циклах. По вреждение рецепторов, как и повреждение мембран, вносит свой вклад в нарушение обмена веществ в клетках за счет сдвига ионного равновесия и изменения водных секторов во вне– и внутриклеточных пространствах. С нарушением ионного баланса связывают осложнения хронического алкоголизма и клинические состояния, возникающие при ААС, такие как судороги, дегидратация, демиелинирующие процессы в нейронах и др. Тяжелое течение ААС, осложняющееся делирием, также объясняют изменением транспорта ионов и аутокоидов через мембраны нейронов (Wettering et. al., 1994).
Продолжение приема спиртных напитков сопровождается стабилизацией дисбаланса центральных возбуждающих и тормозных нейротрансмиттерных систем, однако постепенно плотность НМДА-рецепторов и их чувствительность к глютамату-медиатору увеличиваются. Если в нормальном состоянии НМДА-рецепторы обеспечивают некоторые пластические процессы в нейронах головного мозга (Tabakoff, 1994), то при увеличении их количества и активности могут возникать нежелательные явления, такие как нарушения когнитивных функций вплоть до тяжелого их поражения. Порой могут иметь место такие жизнеопасные нарушения, как ишемия мозга и судороги.
Действие метаболитов этилового алкоголя
Выделенный метаболический аспект токсического действия этилового алкоголя связан с его биотрансформацией, продукты которой являются «четвертой силой», разрушающей клетки изнутри.
Этанол блокирует гексокиназную ферментную систему, которая обеспечивает проникновение глюкозы в клетки и переход ее в глюкозо-6-фосфат. Глюкозу потребляют клетки всех тканей, поэтому при ее недостатке возникает гипогликемия и создаются предпосылки для снижения интенсивности гликолиза и цикла Кребса. Головной мозг и эритроциты особенно чувствительны к гипогликемии, поэтому в организме человека существуют дополнительные источники получения глюкозы на случай чрезвычайной ситуации. Такими источниками являются депо гликогена в печени и ресинтез глюкозы из ее предшественников: аминокислот, лактата и глицерина, которые имеются в каждой группе клеток. Подчеркнем, что все же главным источником поступления глюкозы в организм является пища.
Метаболические последствия в виде гипогликемии, возникающей даже после умеренного однократного приема этилового спирта, частично компенсируются перечисленными биохимическими механизмами. Острая интоксикация этанолом вызывает «биохимическую бурю», а частое употребление спиртных напитков можно сравнить с постоянно действующей чрезвычайной ситуацией для промежуточного обмена веществ.
Во-первых, принятая с пищей глюкоза не усваивается из-за блокады гексокиназы, «да и есть не особенно хочется», поскольку 1 г этанола поставляет около 7 Ккал (для сравнения: это калораж, затрачиваемый на 150 м пути, если грести по воде сидя в лодке). Однако «алкогольные» калории не аккумулируются в макроэрги-ческих связях и дают только тепло.
Во-вторых, при биотрансформации этилового алкоголя в рассматриваемых условиях дополнительные источники получения глюкозы либо блокируются (как, например, глюконеогенез), либо истощаются (как, например, гликогенолиз). В-третьих, при активации перечисленных метаболических путей гормонами, на фоне постоянного присутствия этанола в плазме крови даже в незначительных количествах, образуются агрессивные субстраты, которые депонируются в печени, миокарде, головном мозге и нарушают их функции.
Представленная цепь событий отражает суть возникающих нарушений только в общих чертах. Более глубокий анализ патохимических нарушений при хронической алкоголизации позволяет выделить их ключевые звенья, на которые могут быть направлены усилия фармакотерапии.
Первым метаболитом этанола является ацетоальдегид, который превосходит по токсичности исходную молекулу. Ацетоальдегид преимущественно образуется в гепатоцитах при участии алкоголь-дегидрогеназ (см. рис. 4).
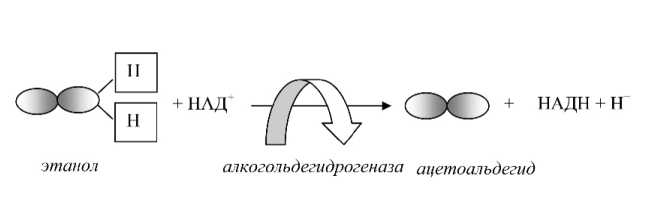
Рис. 4. Образование ацетоальдегида и дефицита НАД+
Условные обозначения:
НАД+ – никотинамиддинуклетотидфосфат;
НАДН – никотинамиддинуклетотидфосфат (восстановленная форма).
Второй протон при восстановлении НАД+ высвобождается в среду. Дефицит НАД+ и одновременное накопление ионов водорода создают условия для развития ацидоза.
Нужно сказать, что все дегидрогеназы переносят электроны и протоны, образующиеся в окислительно-восстановительных реакциях, поэтому они нуждаются в ко-ферментах, т. е. в промежуточных переносчиках функциональных групп. Наиболее важным из них является НАД+-амидникотиновой кислоты, который передает восстановительный эквивалент в дыхательную цепь и участвует в аккумуляции энергии в макроэргах. В результате образования ацетоальдегида НАД+ восстанавливается в НАДН + Н+3, таким образом в ходе реакции биодеградации этилового спирта НАД+ потребляется.
Далее из ацетоальдегида образуется уксусная кислота. Реакция ее образования также протекает с участием альдегиддегидрогеназ, что означает повторное потребление НАД+.
Следовательно, в результате двух метаболических реакций, которые претерпевает этанол в клетках печени, дважды снижается концентрация НАД+.
Дефицит НАД+ сопровождается: блокадой глюконеогенеза; замедлением гликолиза и цикла Кребса; остановкой бета-окисления и отложением жира в печени; изменением поляризации клеточных мембран различных органов за счет сдвига соотношения НАДН/ НАД+.
В результате этого углубляется гипогликемия и возникает дефицит энергии. Клиническими эквивалентами этих изменений являются: слабость, разбитость, «плохое самочувствие» в целом, чувство тяжести в области сердца, головная боль, озноб, гипергидроз, нарушение работы желудочно-кишечного тракта.
С целью компенсации уровня глюкозы плазмы крови распадается гликоген печени. Однако его запасы даже у здоровых людей невелики. Всего лишь на несколько часов гликогенолиз покрывает расход глюкозы нейронами мозга и эритроцитами крови (самыми энергоемкими потребителями), после чего его депо истощаются.
У голодных людей, у лиц, находящихся на диете, у больных диабетом, принимающих гипогликемические средства (за исключением толбутамида), у пациентов, принимающих гепатотоксичные препараты (парацетамол, изониазид, альдомет, фенотиазины и т. д.), бета-адреноблокаторы или средства для снижения массы тела, а также при повторном приеме алкоголя, – запасы гликогена в печени снижены, поэтому гипогликемия у них развивается стремительно, является тяжелой и длительной (Ионин М. Л., 1998). У алкоголиков запасы гликогена в печени снижены почти до нуля.
У здоровых, сытых и трезвых людей существует мощный механизм защиты от гипогликемии. Это синтез глюкозы de novo в печени и в почках. Он осуществляется из глюкогенных аминокислот, лактата и глицерина. Этот процесс называется глюконеогенезом, и его мощность составляет до 250 г глюкозы в сутки. Подчеркнем, что глюконеогенез является самым главным источником питания нейронов и эритроцитов, которые полностью зависят от постоянного интенсивного снабжения глюкозой. Однако для осуществления глюконеогенеза необходим НАД+, запасы которого у лиц, систематически употребляющих спиртное, истрачены на биотрансформацию этилового алкоголя. В этих условиях НАД+ отчуждается от других, порой «недублируемых», биохимических реакций (например, из реакций электронотранспортной цепи флавопротеинов), и они «останавливаются». Это усугубляет дефицит энергии и создает основу для гипоксии различных тканей.
Гликолиз должен осуществляться любой ценой, иначе клетки погибнут. Однако продолжающийся прием спиртного нарушает декарбоксилирование кетокислот и замыкает гликолиз на выходе. Тогда источником образования энергии становятся жиры (триацилглицериды). Они распадаются на жирные кислоты и глицерин. У человека жирные кислоты не могут трансформироваться в глюкозу, они поступают в кровь в неэстерифицированном состоянии (НЭЖК). Жирные кислоты – биохимически агрессивные субстраты – являются источником образования свободных радикалов, стимулируют высвобождение биологически активных веществ, поэтому существует путь их быстрой утилизации в печени, где они превращаются в ацетилкоэнзим-А. Концентрация последнего в печени резко увеличивается, его избыток конденсируется в ацето-уксусную кислоту, из которой образуются 3-гидроксибутират и ацетон, т. е. кетоновые тела. Возникают предпосылки для развития алкогольного кетоацидоза, опасного, рецидивирующего состояния, несвоевременное лечение которого может приводить к летальному исходу.
В свою очередь глицерин также не может трансформироваться в глюкозу (как это происходит у нормальных людей), поскольку этот процесс также катализируется дегидрогеназой, для работы которой также нужен НАД+. В этих условиях в печени вновь синтезируются нейтральные жиры, которые депонируются в гепатоцитах и создают предпосылки для развития их жировой дистрофии.
Таким образом, в условиях гипогликемии, спровоцированной этанолом, «сгорания жиров в пламени углеводов» не происходит, из них образуются кетоновые тела и депозиты жира в печени, ее границы увеличиваются, а функции угасают. Синтетические процессы, происходящие в ней в нормальном состоянии, изменяются, снижается антитоксическая функция печени, что приводит к появлению в крови большого количества аутокоидов, способных вызывать различные нарушения в организме больного.
В попытке повысить уровень глюкозы в плазме крови увеличивается содержание гормонов, регулирующих ее концентрацию, таких как кортизол, глюкагон, гормон роста, адреналин, и снижается секреция инсулина. Гормональные сдвиги практически не влияют на уровень глюкозы, так как ее неоткуда взять. Однако они усиливают липолиз, стимулируют дальнейшее отложение жира в печени и предоставляют материал для дальнейшего образования кетонов.
Сдвиг равновесия НАДН+Н+/НАД+ влево изменяет поляризацию мембран клеток. Потенциал клеточных мембран становится более негативным по сравнению с исходным потенциалом, что еще больше нарушает транспорт ионов через них. Страдают функции транспортных насосов, в том числе тех из них, которые призваны эвакуировать отработанные продукты обмена веществ. Снижение интенсивности обменного транспорта в системе К+/Н+ (антипорт «калий – водород») сопровождается накоплением избытка ионов водорода в клетках. К кетонемическому ацидозу присоединяется внутриклеточный метаболический ацидоз.
В совокупности все это приводит к срыву функции органов, который проявляется в соответствии с их физиологической ролью (нарушения ритма сердца, декомпенсация функций печени, органические дисфункции головного мозга ит. д.).
Однако представленная патохимическая драма не исчерпывается явлениями, связанными с дефицитом НАД+. Сами метаболиты этанола – ацетоальдегид и уксусная кислота, образующиеся при его биотрансформации, – завершают ее финал. Несмотря на то, что ацетоальдегид вырабатывается эндогенным путем во многих биохимических реакциях, превышение его концентрации в головном мозге оказывает выраженное нейротропное и гепато-токсическое действия (Зиматкин С. М., 1993). Ацетоальдегид необратимо связывает белки синаптосом в трофотропных системах ЦНС (например, в ГАМКергических), влияет на продукцию эндогенных опиатов, угнетает транспортные АТФазы мембран нейронов головного мозга (Багров А. Я., 1996), наконец, ацетоальдегид угнетает синтез ацетилхолина – главного функционального антагониста катехоламинергических (возбуждающих) систем головного мозга, индуцирует цитохром СУР 2Е1.
Физиологические пределы колебаний концентрации ацетоальдегида очень малы, его избыток должен немедленно утилизироваться в биохимических реакциях. Однако некоторые продукты биотрансформации ацетальдегида также оказывают повреждающее действие на ткани: например, при деградации ацетальдегида ксантиноксидазой или альдегидоксидазой образуются свободные радикалы. Связывание ацетальдегида с цистеином или глютатионом вызывает увеличение их оборота и потерю пула серосодержащих аминокислот (что также сопровождается накоплением пероксидов и повреждением митохондрий). Длительная алкоголизация и накопление ацетальдегида вызывают снижение пула эндогенного метионина и его активной формы S-аденозилметионина, который участвует в синтезе глютатиона (S-аденозилметионин является наиболее важным агентом в реакциях трансметилирования, которые обеспечивают пластический обмен веществ).
Вместе с тем ацетоальдегид способен усиливать активность ионотропных глицинергических рецепторов (Mascia, 2001). Продукты конденсации ацетоальдегида с норадреналином, серотонином, эндорфинами, такие, как сальсолинол, гармалин и другие, способны конкурентно взаимодействовать с рецепторами указанных лигандов и изменять характер нейро– и котрансмиссии в головном мозге. Возникает хаос в рецепторконтролируемых ионных каналах мембран нейронов.
Это обстоятельство влияет на многие системные процессы, включая функции ВНД, такие как мышление, эмоционально-волевые и другие реакции.
В последние годы установлено, что ацетоальдегид является мощным стимулятором продукции «медиаторов воспаления» интерлейкиновой (IL) группы цитоксинов: IL-6 (фактора некроза опухолей альфа), IL-8 и IL-1, локальные и системные эффекты которых связывают с прямым гепатоцитотоксическим действием, угнетением факторов роста, нейро– и гемопоэза (см. действие на органы), и с развитием демиелинизирующих процессов в головном мозге (Rothwell, 1995).
Уксусная кислота также агрессивна, и поэтому существует механизм ее быстрой утилизации при переходе в ацетилкоэнзим-А, который происходит в митохондриях печени. Реакция его образования высокоэнергетична и сопряжена с экзоэргическими процессами (после приема спиртного всегда становится жарко).
При хроническом алкоголизме метаболический переход ацетальдегида в ацетат снижается. Избыток ацетальдегида в полной мере проявляет представленные выше виды действия.
Еще раз подчеркнем, что избыток ацетилкоэнзима-А блокирует бета-окисление жирных кислот, а также усиливает синтез холестерина и тормозит скорость парциальных реакций цикла Кребса. Это способствует еще большему депонированию жира в печени.
Наряду с дегидрогеназами в биотрансформации этанола принимают участие каталаза и стимулированная этанолом микросомальная оксидаза (система МЭОС с участием цитохромов Р-450). Эти пути биотрансформации этанола «включаются» по достижении его уровня в плазме крови, – в среднем от 1‰ (100 мг%), и они служат способом дополнительной элиминации этого яда из ор га низ ма.
Негативные последствия биотрансформации этанола в системе МЭОС включают:
дополнительное образование ацетоальдегида;
деградацию ксенобиотиков и лекарственных препаратов;
продукцию свободных радикалов и усиление ПОЛ;
избыточный синтез коллагена;
образование аутоантител к продуктам деградации этанола.
При сформированной зависимости от этанола система МЭОС работает наравне с дегидрогеназной системой, увеличивая образование продуктов биотрансформации этилового спирта, т. е. биохимические нарушения в организме усугубляются.
Недегидрогеназная (неокислительная) биодеградация этанола приводит к образованию алкогольных эфиров жирных кислот – биологически активных метаболитов, которые появляются в плазме крови и во внутренних органах при длительной алкоголизации субъекта. Эти вещества оказывают токсическое действие на клетки органов, в которых они образуются.
Чем дольше длится алкоголизация, тем более глубокими являются представленные патохимические нарушения: нарастает функциональная недостаточность различных органов, т. е. возникает полиорганная недостаточность.
Поражения различных систем организма, возникающие при систематическом употреблении этилового алкоголя
Первичный контакт яда с организмом человека происходит на уровне ЖКТ, однако то действие, которое алкоголь оказывает на человека, к нему привычного, принципиально отличается от того действия, которое испытывают люди непьющие или мало употребляющие спиртные напитки. Это умозаключение было впервые высказано J. Pringsheim (1908), который показал, что у алкоголиков спиртные напитки всасываются и разрушаются быстрее, чем это происходит у непьющих людей. Это была одна из первых попыток, предложенных для объяснения явления толерантности к алкоголю, и в те годы было трудно предположить, какие глубокие нарушения гомеостаза у пьющего человека скрываются под вершиной этого «айсберга».
Все изменения, возникающие в органах при хронической интоксикации этанолом, взаимосвязаны друг с другом и происходят из-за действия этилового алкоголя на мембраны, рецепторы и обмен веществ, протекающий в клетках. Между тем в каждой физиологической системе имеются особенности этих изменений, обусловленные выполняемой ею функцией.
Действие на ЖКТ
Здесь следует выделить два фактора, которые влияют на токсикодинамику этанола при хронической интоксикации. В желудке начинается пресистемный метаболизм этанола (его разрушение алкогольдегидрогеназой, которая вырабатывается микробной флорой желудка) и всасывание этанола и ацетоальдегида.
При гастрите и язвенной болезни активность алкогольдегидрогеназы слизистой оболочки желудка увеличена, поэтому у таких лиц прием спиртного сопровождается усилением образования ацетальдегида. Могут возникать осложнения, связанные с его действием (лактацидемия, ацетатемия, выраженные вегетативные реакции, тяжелые алкогольтетурамовые реакции, возможно, и более быстрое развитие привыкания).
Заболевания желудка (болезнь Мэллори – Вейса, синдром Рэндю – Ослера, состояния после его резекции) снижают активность алкогольдегидрогеназы и интенсивность пресистемного метаболизма этанола. В результате эффект опьянения происходит от меньших количеств спиртного.
Препараты, угнетающие активность алкогольдегидрогеназы желудка (аспирин, парацетамол, Н-2-гистаминоблокаторы), замедляют биотрансформацию этанола и снижают токсическое действие, обусловленное ацетальдегидом, однако способствуют усилению опьянения.
Средние показатели активности алкогольдегидрогеназы у женщин вдвое ниже по сравнению с мужчинами. Поэтому при потреблении одинаковых количеств спиртного в плазме крови женщин концентрация этанола нарастает быстрее (иными словами, женщины пьянеют быстрее мужчин).
Частый прямой контакт этанола со слизистыми оболочками желудка, тощей и тонкой кишки вызывает в них дистрофические процессы: отек клеточных элементов, эксфолиацию, нарушение микроциркуляции, атрофию слизистых оболочек и образование язв. Эрозивный гастрит часто является причиной кровотечений из верхних отделов ЖКТ, которые возникают у 20 % хронических алкоголиков (Lawrence et al., 1996). Наиболее уязвимы дистальная часть 12-перстной кишки и проксимальная часть тощей кишки, поскольку в этих отделах равновесные концентрации этанола (в разделах кровь/ткань) удерживаются более длительно, по сравнению с другими отделами кишечника. Морфологические изменения слизистой оболочки сопровождаются нарушением пристеночного пищеварения, всасывания глюкозы, тиамина, фолиевой кислоты, цианкобаламина и, особенно, метионина, что является долнительным фактором в нарушении обмена веществ и развитии осложнений алкоголизма.
В указанных отделах кишечника локализованы гипофиз-независимые нейроэндокринные клетки, секретирующие в кровь вещества, аналоги которых в головном мозге выполняют медиаторные и другие функции. При иммунохимической оценке секреторной активности биоптатов дистального отдела нисходящей части 12-перстной кишки у здоровых непьющих добровольцев и пациентов с низким уровнем потребления спиртного (менее 40 г/неделю) была выявлена большая плотность рецепторов холецистокинина, галанина, ингибиторного пептида желудка, глюкагона, мотилина, нейропептида Y, питуитарного аденилатциклазо-стимулирующего пептида, секретина, серотонина, соматостатина, субстанции Р, ВИП-гормона, чем в биоптатах пьющих людей (Hauge et al., 2001).
Исследование механизмов химической зависимости по составляющим APUD-систему аутокоидам является весьма перспективным в силу доступности и возможности визуального, морфологического и гистохимического исследования этих отделов ЖКТ. Используя в качестве инструмента теорию тождества регуляторных систем «на входе и выходе» (в частности, холинореактивных систем (Денисенко П. П., 1982)) применительно к аутокоидам APUD-системы и рассматривая абстинентные синдромы, возникающие при отмене героина, клофелина, этанола, бета-адрено-блокаторов и т. д., в виде типовых патологических процессов, в рабочей гипотезе «о наркомании – болезни кишечника?» мы высказали предположение о возможной маркерной роли аутокоидов APUD-системы в установлении предрасположенности к действию наркотических веществ (Афанасьев В. В., с соавт., 1986).
Следует подчеркнуть, что всасывание этанола в желудке контролируется холинореактивными системами через М-холинорецепторы, причем участвуют в этом процессе все их подтипы, кроме М-1-рецепторов (Kinoshita, 2001).
При алкоголизации эрозируется слизистая оболочка желудка и тонкого кишечника. В совокупности с изменениями свертывающих свойств крови это обстоятельство может приводить к желудочно-кишечным кровотечениям.
Алкоголь также проявляет панкреатотоксическое действие. Обострение хронического панкреатита особенно часто возникает при приеме спиртного на фоне (или совместно) с лекарственными средствами, оказывающими панкреатотоксическое действие и при употреблении сахара («закусить конфеткой»).
Действие на ЦНС
Изменения в ЦНС являются следствием патохимических процессов, индуцированных систематическим приемом этилового алкоголя. Биохимические нарушения промежуточного, белкового и других видов обмена веществ, мембранотоксические эффекты, дисрегуляция рецепторного аппарата клеток и их ферментных систем, образование большого количества биологически агрессивных веществ в клетках ЦНС и их матриксе – играют роль в нейроадаптации нейронов головного мозга, нейрофармакологической сущностью которой является стойкое преобладание активности возбуждающих структур в ЦНС над тормозными, вплоть до глубокого угнетения функциональной активности послед них.
Исследованиями Э. А. Костандова с соавторами (1981) было установлено, что экспозиция этанолом сопровождается большим угнетением корковой активности правого полушария головного мозга. Авторы обратили внимание на сходство психопатологической продукции, возникающей при очаговых органических поражениях правого полушария головного мозга и хронической алкоголизации, таких как беззаботность, беспечность, благодушие, своеобразный юмор («алкогольный» юмор), нарушение критики и осознания своего заболевания и его последствий и другие признаки.
У лиц, хронически употребляющих этанол, практически во всех отделах мозга в той или иной степени имеются структурно-функциональные изменения: во фронтальной коре большого мозга, медиальных и темпоральных долях, гиппокампе, диэнцефалоне, переднем мозге. В меньшей степени задействованы стрио-паллидарные структуры. В перечисленных отделах мозга снижается региональный кровоток, возникают атрофические процессы, происходит редукция астроцитной глии и олигодендроцитов. Существенно снижается общий вес мозговой ткани (по данным 8735 аутопсий вес мозга алкоголиков в среднем на 37 г меньше веса мозга у лиц без алкогольной патологии (Torvik et al., 1982)). Нейро-патологические изменения мозга при аутопсии алкоголиков-муж-чин в возрасте от 30 до 64 лет выявлены в 67,7 % случаев (представлены результаты 194 аутопсий). Наиболее частыми из них были: энцефалопатия Вернике (14,2 %), церебральная атрофия (37 %), демиелинизация волокон Варолиевого моста (2 %), печеночная энцефалопатия (1 %), причем в 28 % случаев регистрировались сочетанные поражения (Skullerud et al., 1991).
Нарушаются когнитивные функции мозга, страдает память, концентрация внимания, изменяется эмоционально-волевая деятельность и тонкие социальные связи. Возникают внешние и внутренние конфликты, формируются варианты подкрепления, постепенно структурируются мотивация и элементы аддиктивного поведения. Между девиантным поведением детей, рожденных от родителей-алкоголиков, и электровозбудимостью нейронов фронтальной коры их головного мозга существует взаимосвязь, прослеживающаяся в поколениях. Корреляция между предрасположенностью таких подростков к алкоголизму и снижением амплитуды (и скрытого периода) поздних волн, вызванной электрической активностью головного мозга (так называемая волна Р300), у них настолько высока, что позволяет выделять изменение Р300 в качестве маркера этого заболевания.
Длительное и частое употребление этанола провоцирует развитие цереброваскулярной патологии. В ближайшее время после приема большого количества спиртного в среднем на сутки усиливается тромбообразование, которое может приводить к острому нарушению мозгового кровообращения. Спустя несколько суток после приема большого количества спиртного увеличивается частота геморрагических инсультов (Taylor, 1982), причем у женщин субарахноидальное кровоизлияние встречается чаще, чем у мужчин.
В основе геморрагических нарушений лежит волнообразное изменение синтеза биологических веществ в системе арахидоновой кислоты, возникающее при чередовании абстиненции и интоксикации, что приводит к волнообразному изменению реологических свойств крови. Другой причиной является либерализация этанолом определенной группы цитокинов в клетках оболочек головного мозга и его нейронах. В интервале 24–48 ч цитокины (особенно IL– 1b– и g-интерферон) увеличивают продукцию NO в этих клетках, что вызывает вазодилатацию (Shih, 2001).
В последние годы большое значение в развитии геморрагических инсультов при «алкогольной» болезни отводят гипомагнигистии и гипомагниемии. Раннее выраженное и длительное снижение концентрации этого иона в телах нейронов, особенно при сочетанной патологии (например, ААС и ЧМТ) сопровождается вазоспазмом церебральных сосудов, снижением креатинфосфата в клетках ЦНС, падением рН и редукцией фосфорилирующего потенциала цитозоля нейронов (отношение креатинфосфата к неорганическому фосфору). Эти клинические наблюдения были подтверждены интересным экспериментом Altura (1999) на животных, с использованием имплантатов мини-помпы для перфузии желудочков мозга раствором этанола, выполненных по типу ставших уже историческими опытов Feldberg (1956). Перфузия мозга 30 %-ным раствором этанола вызывала 30 %-ное снижение концентрации внутриклеточного магния, которое удерживалось вплоть до 14-го дня после окончания перфузии и сопровождалось падением уровня креатинфосфата и фосфорилирующего потенциала цитозоля на 15 и 40 %, соответственно, причем впоследствии у животных повышалась чувствительность к токсическому действию «малых» доз алкоголя, введение которых вызывало у них геморрагический инсульт.
Активация НМДА-рецепторов сопровождается гиперкалцигистидией нейронов головного мозга. Ионы кальция, являясь функциональными антагонистами ионов магния, играют большую роль в возникновении цереброваскулярных осложнений (Одинак М. М. с соавт., 2001), в том числе возникающих при ААС.
Имеются сведения о том, что в дозах до 0,5 мл/кг массы тела этанол снижает аггрегацию тромбоцитов, тем самым влияя на тромбоцитарно-сосудистый гомеостаз.
Прогрессирование хронической интоксикации приводит к энцефалопатической стадии алкогольной болезни. Алкогольная энцефалопатия, дементность, мозжечковая дегенерация, печеночная энцефалопатия часто необратимы.
Важно отметить, что некоторые патохимические процессы, происходящие в тканях мозга, могут усугубляться при проведении инфузионной терапии лицам, страдающим хроническим алкоголизмом, и приводить к осложнениям, порой жизнеопасным. Это обстоятельство следует учитывать, особенно при современном распространении частной службы «обрыва запоев». Так, по данным Ленинградского областного бюро судебно-медицинской экспертизы, в стационарах Ленинградской области летальность при хроническом алкоголизме и его осложнениях составила 57 % от общего числа лиц, умерших от острых отравлений (Афанасьев В. В. с со авт., 2000).
Клинические признаки поражения нервной системы хорошо заметны: статическая атаксия (даже в трезвом виде), тремор рук, языка, гиперемия лица, повышение (или снижение) сухожильных рефлексов, полиневропатии.
Многие патологические явления, возникающие при хронической интоксикации у человека, можно моделировать на животных. В исследовании Т. Н. Саватеевой с соавторами (2002) развитие алкогольной энцефалопатии возникало в течение 23 дней затравки крыс 15 %-ным раствором этилового алкоголя. Клиническая картина этого состояния сопровождалась замедлением скорости обработки информации, нарушением условно-рефлекторной деятельности животных, которые были обусловлены первичной атрофией нервных клеток и расширением желудочков мозга, снижением электросубкортикограммы, особенно выраженным в лобных отделах головного мозга. Несмотря на кратковременность экспозиции алкоголем у животных возникали изменения биохимических показателей крови, свидетельствовавшие об оксидативном стрессе, вызванном систематическим приемом спиртного, и снижался уровень клеточного звена иммунной системы.
Хронический алкоголизм является второй, наиболее распространенной после диабета причиной, вызывающей поражение периферической нервной системы. Токсическая полинейропатия (моторная и сенсорная) первоначально проявляется в виде выпадения проприоцептивного чувства (парастезии по типу «перчаток», онемение конечностей, чувство дискомфорта в мышцах), затем возникают боли в мышцах, слабость мышц и их атрофия, особенно заметная в нижних конечностях. Болевой синдром может быть настолько выраженным, что не купируется нестероидными противовоспалительными средствами. В его основе лежит аксональная дегенерация нервных проводников, обусловленная действием алкоголя и его метаболитов.
Действие на сердечно-сосудистую систему
Хроническое употребление этанола находится в прямой зависимости с развитием артериальной гипертензии, нарушениями ритма сердца, ишемической болезнью сердца и дилатационной кардиомиопатией. Гипертензия является наиболее ранним проявлением частого употребления спиртного. Систематический, каждодневный прием алкогольных напитков даже в небольших количествах (18 мл этанола/сутки) вызывает стойкое повышение АДсист. и АДдиаст. Гипертензия достаточно ригидна к действию антигипертензивных средств, но она обратима, и после прекращения алкоголизации артериальное давление имеет тенденцию к снижению.
Метаболиты этанола и вещества, высвобождающиеся при его действии, оказывают прямой констрикторный эффект на коронарные артерии и тонус сосудов в целом. Вазоспазм обусловлен катехоламинемией, увеличением в плазме крови концентрации аутокоидов-вазоконстрикторов, среди которых в последние годы особое внимание отводят эндотелину-1 и гомоцистеину. Концентрация эндотелина-1 повышается в плазме крови почти вдвое, достигая максимума через 5 ч после приема спиртного (Kaku et al., 1999), и удерживается в ней длительное время. Умеренная алкоголизация сопровождается увеличением уровня гомоцистеина, образующегося в том числе при биотрансформации катехоламинов.
Гомоцистеин является лигандом-агонистом НМДА-рецепторов и нейротропным путем вызывает кардиоцитотоксический эффект (Bleich. et al., 2000).
Вместе с тем французские авторы на основании данных эпидемиологических исследований отмечают, что прием этанола в умеренных количествах (не более 35–40 г/сутки, в виде вина) снижает частоту ишемии миокарда – так называемый «французский парадокс» (Renaud et al., 1988; Hendriks et al., 2001). Однако даже через месяц после умеренного, но постоянного употребления этанола в миокардиоцитах выявляются отчетливо различимые ультраструктурные изменения в виде деградации митохондрий, липидных включений в цитоплазме и внутриклеточного отека миокардиоцитов (Anon, 1979).
Метаболиты этанола также оказывают прямое токсическое действие на мышцу сердца. Считается, что при систематическом приеме спиртного в дозе более 80 г/сутки на протяжении 10 лет в миокарде возникают необратимые изменения, которые лежат в основе миокардиопатии. Прогредиентно снижается сердечный выброс, развивается хроническая недостаточность сердца (Preedy, 1994). Примерно в эти же сроки может формироваться алкогольная дилатационная (так называемая конгестивная) кардиомиопатия. Ее патогенез изучен недостаточно. Известно только, что в миокардиоцитах образуются алкогольные эфиры жирных кислот вследствие неокислительного пути биотрансформации этанола. Эти вещества откладываются в митохондриях миокардиоцитов и вызывают их разрушение.
Между наличием в миокарде алкогольных эфиров жирных кислот и образованием дилатационной кардиомиопатии выявлена прямая причинно-следственная связь (Beckemeir et al., 1998). Кардиомиопатия проявляется увеличением границ сердца, дилатацией его камер и прогрессирующей некоррегируемой сердечной недостаточностью. Сформированным морфологическим субстратом кардиомиопатии является фиброз миокардиоцитов и их инфильтрация гликопротеином.
Нарушения ритма сердца являются характерными для хронического алкоголизма. Чаще всего они проявляются мерцательной аритмией (у 10 из 14) (Гришкин Ю. Н., 1995; Greenspon, 1983). Известно, что синдром внезапной смерти намного чаще возникает у лиц, принимающих в день более шести условно стандартизованных доз этанола[2], по сравнению с теми, кто пьет меньше (Warramethee, 1992).
При ААС аритмии – наиболее частая причина острой сердечно-сосудистой недостаточности и смерти. Нарушение ритма сердца связывают с синдромом удлиненного Q-T, который провоцирует фибрилляцию желудочков. В свою очередь увеличение интервала Q-T обусловлено длительной алкоголизацией, в ходе которой «скрещиваются» три неблагоприятных явления, каждое из которых способно самостоятельно вызывать нарушения ритма сердца: гипомагниемия, гипокалиемия, адреналинемия и норадреналинемия. Последняя особенно выражена при ААС в интервале 7—24 ч (Carlsson, 1967). В связи с этим необходимо заметить, что фармакотерапия ААС психотропными препаратами может изменять не только тонус вегетативной нервной системы, но и электролитный баланс организма и приводить к нарушениям ритма сердца, даже если психотропные препараты назначают в терапевтических дозах (ЕД50). Замечено, что при хронической алкоголизации возникает так называемая автономная миокардиальная нейропатия (Weise, 1986) с чередованием преобладания адрен– и холинергических влияний, в результате которых может колебаться величина интервала Q-T. Размах этих колебаний становится еще большим при ААС. По данным Otero-Anton (1997), у половины больных ААС регистрируют синдром удлиненного Q-T при поступлении в стационар. Это обстоятельство делает опасным проведение лечения ААС на дому.
Д. М. Табеева с соавторами (1997) установила фазность чередований холин– и адренергических влияний при ААС. Авторы выделили 4 следующих друг за другом периода: выраженная симпатикотония (1—5-й дни ААС), менее выраженная симпатикотония (6—10-й дни), максимальная парасимпатикотония (11—30-й дни) и нормализация автономных проявлений с преобладанием холинергических воздействий (30-й день и далее). Изменение концентраций гистамина, серотонина, ацетилхолина, адреналина и норадреналина при хронической интоксикации этанолом и отмене этанола были продемонстрированы Т. Г. Наимовой (1979) в эксперименте на животных.
Действие на систему дыхания
У большинства больных хроническим алкоголизмом имеется поражение легких. Оно обусловлено патогенетически и тесно связано с нарушением функций печени, снижением иммунитета и непосредственным повреждением ткани легких. Большое значение имеет постоянная элиминация алкоголя легкими. Вследствие прямого раздражающего действия алкоголя и его метаболитов, оказываемого при их выведении из организма, возникает хроническое воспаление участков бронхов, гиперплазия эпителия и усиление нейтрофильной инфильтрации альвеол. Из-за недостаточности функций печени в плазме крови появляются избыточные концентрации биологически активных веществ, которые в нормальных условиях разрушаются печенью. С током крови они попадают в альвеолы и воздействуют на их структурные элементы.
Избыток гистамина увеличивает проницаемость легочных капилляров, блокада катаболизма лейкотриенов в печени приводит к увеличению их содержания в плазме крови и развитию воспалительных реакций в интерстиции легких. Лейкотриены Е-4, В-4 и ациллейкотриен способны длительное время циркулировать в крови и осаждаться в легких, как в фильтре. С увеличением их концентрации в плазме крови и в моче связывают этанол-индуцированную реакцию эндогенной интоксикации. Так, базальная экскреция лейкотриена Е-4 в моче обычно составляет 0,5 нмоль/л, в то время как при хроническом приеме этанола она увеличивается до 1,5 нмоль/л и сохраняется в течение 3–5 ч, после того как концентрация этанола снижается до нуля (Uemura et al., 2000).
В пульмоноцитах снижаются запасы глютатиона, возникает сдвиг в сторону его окисленной формы. Параллельно увеличивается продукция коллагена матриксом альвеоцитов, что закладывает основу для фибротических и обструктивных процессов в них. У пациентов увеличивается процент неработающих альвеол, растет венозное шунтирование крови, понижается SpO2, возникает артериальная гипоксемия. В результате этого страдают газообменные функции легких, развивается застой крови в них, появляется склонность к бронхоспазму и к воспалительным и деструктивным заболеваниям респираторной системы в целом.
Острый респираторный дистресс синдром (ОРДС) при хроническом алкоголизме развивается у 29 % госпитализированных больных с осложненным течением ААС, причем летальность при таком состоянии составила 51 % по сравнению с 14 % среди других групп больных (Назаренко В., 2000).
Острые пневмонии, осложненные инфекционно-токсическим шоком, респираторным дистресс синдромом (Moss, 2000) и отеком легких являются центральным звеном танатогенеза критических состояний, возникающих при ААС.
Действие на систему крови
Следует выделить четыре категории процессов, которые происходят в клетках и плазме крови: влияние на цитокиновые системы, гемопоэз, свертывающие свойства крови и токсификацию плазмы крови.
Изменение цитокиновой регуляции. Цитокины высвобождаются клетками крови и присутствуют по всех органах и тканях. По сравнению с гормонами цитокины контролируют большее количество клеток-мишеней. Цитокины весьма разнообразны: интерлейкины, лимфокины, монокины, хемокины, интерфероны и т. д. Они создают своеобразную регуляторную «сеть» и «перекрывают» действие друг друга. Некоторые цитокины являются антагонистами. Ко многим из них существуют антицитокины. Рецепторы цитокинов локализованы на внешней стороне клеточных мембран и являются специфическими для многих представителей группы. Изменение структуры липидного бислоя нарушает трансформацию цитокинового сигнала за счет снижения аффинности рецепторов к цитокинам, а также из-за нарушения нормального передвижения в бислое их транспортных белков-переносчиков.
Таким образом, хроническая интоксикация этанолом вызывает дисбаланс цитокинового каскада. При этом возникает увеличение удельного веса интерлейкиновой группы цитокинов, особенно в период абстиненции (Gonzalez – Quitela, 1999). Среди последних выделяются «провоспалительные» интерлейкины (IL-1b; IL-6; IL-8; IL-10; IL-12 и др.), увеличение концентрации которых в плазме крови сопровождается ростом IgE, гепатоцитотоксическим и панкреатоцитотоксическим действием, угнетением факторов роста и т. д. При детоксикации концентрация некоторых цитокинов снижается, что создает особую предрасположенность к возникновению инфекционных осложнений в этот период (Maes, 1998). Действительно, на следующие сутки после проведения инфузионной терапии температура тела у некоторых больных (алкоголиков, без заболеваний печени) увеличивается и, в первую очередь, появляются признаки респираторных расстройств. Это явление также может быть связано с тем, что интерлейкины (IL-1) способны активировать цитотоксические Т-клетки. Последние узнают и связывают инфицированные вирусами клетки организма и таким образом оказывают цитотоксическое действие при сопутствующих алкоголизму вирусных заболеваниях (например, гепатоцитотоксическое действие интерлейкинов при гепатитах), так как известно, что вирусные поражения печени обнаруживаются у четверти алкоголиков.
Изменение гемопоэза. Хронической алкоголизации практически всегда сопутствует анемия, эритроциты уменьшаются в размерах, деформируются, приобретают атипичное строение (акантоциты, стоматоциты). Выявленная путем сканирующей электронной микроскопии степень их деформации может указывать на глубину поражения печени (Takashimizu et al., 2000). Такие клетки неполноценны, плохо выполняют свою роль в переносе кислорода и обмене его в тканях, нарушают микроциркуляцию в них. Угнетается образование лимфоцитов, возникает лимфопения, происходит снижение резистентности организма к инфекции. В той или иной степени этанол действует на факторы роста всех ростков кровотворения, деятельность которых во многом регулируется цитокинами.
Изменение свертывающих свойств крови. Сам этанол оказывает дозозависимый эффект на свертывающие свойства крови. Его прием в малых дозах, до 0,5 мг/кг массы тела/сутки («рюмка» водки), сопровождается ингибированием агрегации тромбоцитов. Это действие связано со снижением уровня арахидоновой кислоты и редукцией синтеза тромбоксанов.
Нарушения в системе тромбоцитарно-сосудистого гемостаза («агрегация/дезагрегация») возникают у алкоголиков в 8 раз чаще, по сравнению со здоровыми лицами (Micromedex, Inc., 2000), так как постоянный прием спиртного вызывает изменение фосфолипидного состава биологических мембран и нарушение химических синтезов эйкозаноидов, для которых фосфолипиды являются предшественниками. При употреблении спиртного из расчета 1,5 г/кг массы тела/сутки отмечена повышенная концентрация тромбоксана В(2) в крови и моче больных, сопровождающаяся отчетливым повышением агрегации тромбоцитов и увеличением вязкости крови в ближайшее время после запоя.
Следует отметить, что маркером сформированной зависимости от этанола может служить увеличение тромбоцитарной аденилатциклазы. Специфичность и чувствительность метода составляют 79 и 75 % (Menninger et al., 2000).
Изменение токсификации плазмы крови. Токсификация плазмы крови и эритроцитов, измеренная путем оценки уровня МСиНММ – молекулы средней и низкой молекулярной массы (более 5000 дальтон), является прямо пропорциональной степени клинических расстройств, наблюдаемых при ААС, причем ААС II и III степеней тяжести, по уровню эндотоксемии сопоставим с такими тяжелыми состояниями, как травма и панкреатит (Малахова М. Я., 1993). МСиНММ включают большое число аутокоидов, многие из которых способны высвобождать соединения, оказывающие повреждающее действие на клетки (например, интерлейкин 10), при этом эндотоксин-нейтрализующая способность плазмы крови снижается. Существует мнение, что увеличение токсификации плазмы крови находится в прямой связи с алкогольным поражением печени (Urbascek, 2001).
Действие на печень и обмен веществ
В гепатоцитах снижается интенсивность гликолиза и глюконеогенеза вследствие дефицита глюкозы и НАД. Из-за алиментарного недостатка страдает белковый обмен. По мере прогрессирования алкоголизации возникает дефицит важных активированных метаболитов (УДФ-глюкозы, ЦДФ-холина, S-аденозилметиони-на), осуществляющих биохимические взаимосвязи обмена веществ в клетках различных типов.
Это может приводить к нарушению реакций ацетилирования, фосфорилирования и, особенно, метилирования в гистоновых белках хроматина, непосредственно связанных с ДНК в ядрышках гепатоцитов. В результате снижается синтез белков, понижается способность клеток печени к регенерации, формируются фибротические изменения в гепатоцитах. Ядра клеток теряют способность поставлять в цитоплазму НАД+.
Дефицит некоторых метаболитов и белков мог бы быть покрыт алиментарным путем, однако при алкоголизме практически всегда имеет место нарушение питания либо оно является неправильным. Коррекцию недостатка определенных метаболитов, некоторые из которых можно условно назвать «пищевыми добавками»[3], следует проводить при лечении ААС и при поддерживающей терапии в постабстинентном периоде.
При хронической интоксикации этанолом возникает индукция цитохромов (ферментов системы МЭОС), нарабатывающих ацетоальдегид и свободно-радикальные субстанции, активирующие процессы перекисного окисления липидов. Их активность может повышаться от 4 до 10 раз (Leiber, 1997). Именно эти цитохромы «виновны» в формировании толерантности к этанолу и ко многим лекарственным препаратам, используемым для лечения алкоголизма и сопутствующих ему заболеваний (ожирения, диабета). За счет повышенной активности системы МЭОС происходит ускоренная деградация эндогенных антиоксидантов и других субстратов, образуются токсические метаболиты в результате биотрансформации лекарственных веществ.
В экстрацеллюлярном матриксе нарушается соотношение между составляющими его веществами: коллагеном, адгезивными белками и протеогликанами. Осевой молекулой для этих веществ является гиалуроновая кислота. За счет снижения ее биотрансформации увеличивается концентрация гиалуроновой кислоты в матриксе. Таким образом, создаются предпосылки для формирования септальных фибротических изменений. При этом на апикальных участках гепатоцитов увеличивается количество рецепторов к гиалуроновой кислоте (рецептор СD44), которые могут служить маркерами алкогольного поражения печени (Urashima, 2000).
Даже при умеренной экспозиции этанолом перечисленные явления могут сопровождаться неспецифическими морфологическими изменениями гепатоцитов в виде небольших четко очерченных жировых включений, свидетельствующих о начале их жировой дистрофии. Ультраструктурные изменения гепатоцитов в виде жировых включений наблюдались у здоровых добровольцев при приеме ими 270 г водки за вечер (Robbins S., 1984).
Нарушения митохондриального окисления жиров и изменение их внтуриклеточного перераспределения в гепатоцитах приводят к стеатозу последних. Количество включений растет, размеры печени увеличиваются. Гистологически подтвержденный стеатоз печени (с разрушением митохондрий, деградацией элементов цитоплазматического ретикулума, вакуолизацией гепатоцитов, аккумуляцией жировых включений в них) возникает через 10–12 дней после обильного употребления алкоголя, особенно если оно сопровождалось исключением из диеты углеводов (Rubin, 1967).
Дальнейшее употребление этанола вызывает характерные воспалительные изменения в гепатоцитах. В них образуются «мегамитохондрии», алкогольный гиалин, сателлитоз и фибротические изменения в синусоидах, т. е. морфологические признаки алкогольного гепатита. Его первоначальные клинические признаки неспецифичны. Больные предъявляют жалобы на чувство «тяжести» в правом подреберье, регистрируются субфибрилитет, лейкоцитоз, увеличение активности ферментов. Однако при этом поражение печени может быть существенным, и очередная интоксикация, даже легкой степени, может сопровождаться развитием ее недостаточности. У тучных алкоголиков морфологические признаки поражения печени появляются в 2–3 раза быстрее, чем у лиц пониженного питания. Тучность – предиктор поражения печени при хроническом алкоголизме (Bunout, 1999).
Следует отметить, что алкогольный гиалин (или тельца Мэллори) обнаруживают при болезни Вильсона – Коновалова, первичном билиарном циррозе печени, фокальной узелковой гиперплазии, гепатоцеллюлярной аденокарциноме, т. е. при всех вариантах нарушений цитоархитектоники гепатоцитов, сопровождающихся их дегенерацией. Важно помнить, что при алкогольном гепатите тельца Мэллори могут резорбироваться после прекращения приема этанола. В среднем этот период занимает 6—12 недель (Mitros, 1999). Поэтому изменение размеров митохондрий при гистологическом исследовании печени может свидетельствовать об обильном употребления этанола на протяжении последнего месяца до момента морфологического исследования биоптатов печени (Mitros, 1999).
Длительная экспозиция этанолом приводит к циррозу печени. «Алкогольный цирроз» первоначально сопровождается перивенулярным, а позднее – перцеллюлярным прогрессирующим фиброзом, причем у женщин он развивается быстрее и от меньших количеств спиртного, по сравнению с мужчинами (Leiber, 1991).
В целом считают, что жировая дистрофия печени является наиболее частой и типичной формой ее поражения при алкоголизме, она развивается у 90 % лиц, систематически употребляющих спиртное (Mitros, 1999). Далее следует алкогольный гепатит, который регистрируется у 10–35 % лиц с зависимостью к этанолу (Цинзерлинг В.В., 1998) и алкогольный цирроз печени, который регистрируется в 10 % наблюдений (Mitros, 1999).
Специфическим маркером алкогольного гепатита может служить полипептидный специфический антиген (TPS), используемый для диагностики опухолевого роста.
Существует корреляция между увеличением концентрации этого полипептида в ткани печени и дегенерацией телец Мэллори. При алкогольном гепатите концентрация TPS увеличивается до 1486 Ед/л (от 176 до 5023 Ед/л; n = 77), при стеатозе она составляет 106 Ед/л (от 41 до 221 Ед/л; n = 77), нормальные значения показателя у здоровых людей составляют 79 Ед/л, с колебаниями от 19 до 235 Ед/л (Gonzalez – Quintera et al., 2000).
У 25 % алкоголиков с поражением печени выявляют антитела к гепатиту С (Leiber, 2000).
У больных с наследственным (аутосомно-доминантным) дефектом активности ферментов, участвующих в синтезе гема, может возникать острая (печеночная) порфирия, спровоцированная хроническим приемом алкоголя. Это потенциально опасное заболевание, которое часто не диагностируется, поскольку порфирия имеет черты сходства с клинической картиной ААС, осложненной периферической мотосенсорной полиневропатией или энцефалопатией. Факторами риска в возникновении острой порфирии считают низкоуглеводную диету, естественные гормональные перестройки организма, прием для купирования ААС лекарственных веществ, таких как барбитураты, антидепрессанты, диуретики, антигистаминные средства и другие препараты. Накопление предшественников гема (d-аминолевулиновой кислоты) и порфобилиногена способно вызывать аксональную дегенерацию, блокировать АТФазные ферменты в мембранах нейронов и выполнять роль медиатора в ГАМКергических системах ЦНС в условиях его дефицита. Совокупность таких эффектов может приводить к энцефалопатии (центральное действие) и полиневропатии (периферическое действие), а также судорожным синдромам и делириозным расстройствам, что существенно осложняет дифференциальную диагностику и лечение ААС. В связи с этим следует подчеркнуть, что спирт этиловый в дозозависимом порядке активирует каспазу-3 и сфигномиелиназу гепатоцитов, сигнальные функции которых обусловливают пусковые реакции апоптоза во многих типах клеток (Liu, 2000) и тем самым может приводить к угасанию не только функций гепатоцитов, но и клеток других органов.
Голод, неправильное питание, авитаминозы В, С, Е, а также диеты с целью снижения массы тела вызывают редукцию запасов гликогеновых депо и естественных антиоксидантов в печени. Эти состояния делают печень чрезвычайно чувствительной даже к небольшим количествам спиртного. Заметим, что в экспериментальной практике при изучении острого и хронического действия этанола исследователи предъявляют очень жесткие требования к отбору и содержанию животных (диета, световой день, толерантность и т. д.), иначе результаты эксперимента подвергаются сомнению.
Действие на орган зрения
В фотохимической реакции передачи светового сигнала родопсином участвуют глютаматергические системы и G-белки, ограничивающие активность этого сигнала. В темноте зрительная клетка беспрепятственно выбрасывает медиатор глютамат в синаптическую щель, что, возможно, сопровождается адаптированностью рецептора к его действию в режиме сумерки/свет. Анкетирование алкоголиков, проведенное нами совместно с И. В. Комаровым (2000), показало, что подавляющее большинство из них чувствуют себя в сумерках комфортнее как во время приема алкоголя, так и в период абстиненции. Возможно, по этой причине поражение зрения у них – явление достаточно редкое. Однако следует отметить, что у части субъектов возникает специфическое нарушение цветоощущения – тританопия – окрашивание окружающих предметов в желтовато-голубоватые тона (Russel, 1980). Очередная интоксикация этанолом может сопровождаться нистагмом, оценка которого (по Ташену) может оказаться полезной, при отсутствии возможности химико-токсикологического исследования биосред, с целью определения ориентировочного уровня этанола в плазме крови. Легкий нистагм появляется при содержании этанола в плазме крови от 0,6 до 0,8 %, его продолжительность составляет 9—14 с (оценивается полуколичественно в виде +) (Бережной Р. В., 1980). При концентрации 0,8–1,2 % продолжительность нистагма составляет 14–19 с (++), при концентрации 1,25—2,0 % продолжительность нистагма составляет более 20 с (+++). Инъецированность сосудов конъюнктивы часто встречается при осмотре больных.
Действие на костно-мышечную систему
Поражение костной и мышечной тканей отражают системные нарушения белкового обмена при хроническом алкоголизме. Нарушение обмена коллагенов, составляющих до 25 % от общего количества белка в организме, приводит к снижению их количества в костной ткани. Возникает остеопороз костей, который часто приводит к их переломам, в том числе при ЧМТ. Вместе с тем замечено, что у женщин в менопаузе прием умеренных количеств этанола увеличивает плотность костной ткани (Laitinen, 1991). В мышечной ткани могут возникать поражения актомиозинового комплекса, плазматических мембран миоцитов, межмышечной соединительной ткани и мышечных частей синапсов. Алкогольная миопатия развивается независимо от алкогольной нейропатии и является самостоятельным заболеванием, которое проявляется дистрофией мышечных волокон и наблюдается у 2/3 больных хроническим алкоголизмом. Это заболевание проявляется слабостью, дряблостью мышц и снижением массы мышечных групп плечевого пояса. Особенно заметной является потеря мышечной массы нижних конечностей, которая может достигать 30 % и более от исходной массы мышц. Для алкогольной миопатии характерны боли в мышцах, часто в сочетании с их локальным отеком. Иногда отмечаются поражение гладких мышечных волокон и связанная с этим дисфагия (вследствие поражения мышц пищевода). Алкоголь и ацетоальдегид ингибируют активность мышечной РНК и нарушают синтез белка в мышцах.
В настоящее время миопатии подразделяют по типам нарушенного синтеза белка, который контролируется геном или группой генов. Многие гены картированы, что дало возможность установить продукт гена и определить первичную зону биохимического поражения (дистрофинопатии, ламининопатии, эмиринопатии и т. д.). Для уточнения биохимического субстрата миопатии используют молекулярно-генетические методы (обнаружение мутантного гена) и иммуногистохимическое (субстратное) определение антител в биоптатах мышц.
Важными факторами в развитии алкогольной миопатии считают гипокалиемию и гипофосфатемию, причем, в отличие от рабдомиолиза, при алкогольной миопатии нет резкого увеличения креатинфосфокиназы крови и миоглобина в моче. Морфологическими признаками алкогольной миопатии являются некротические изменения в мышечных волокнах I типа (низкое содержание миозиновой АТФазы) и атрофические процессы в волокнах II типа (высокое содержание миозиновой АТФазы).
Действие на эндокринную систему
Гипогликемия развивается из-за алиментарных причин и блокады глюконеогенеза, наиболее часто – в интервале 2—10 ч после приема спиртных напитков. У алкоголиков гипогликемия может сохраняться длительно, в течение 6 и более часов. Для ее развития бывает достаточно принять 0,7 г/кг этанола (что в среднем создает концентрацию в плазме крови в пределах 1‰ (100 мг%), достаточную для «остановки» глюконеогенеза). При длительной гипогликемии стремительно развиваются необратимые изменения в нейронах, вплоть до их некрозов. Последующее восстановление уровня глюкозы крови не приводит к успеху.
Летальность при индуцированной этанолом гипогликемии составляет 10 % (Service, 1995), гипогликемическая кома у алкоголиков развивается в 33 % случаев (Klatt, 1988).
Нарушения гормонального статуса в наибольшей степени изучены в системе гипоталамус – гипофиз – гонады (ГГГ). Дисбаланс в системе ГГГ в абстинентном периоде проявляется несоответствием между высоким уровнем кортизола плазмы крови и низкой концентрацией нейростероидов (дигидроэпиандростеро-на) в ней, что свидетельствует о снижении стресс-протективных возможностей пациента и может отражать степень имеющихся у него депрессивных расстройств (Heinz, 1999).
Стимуляция диуреза у больного при проведении детоксикации (чаще в режиме «обрыва запоя») и «вымывание» стресс-протективных аутокоидов требуют восстановления их количества в ближайшее время, после проведения процедуры, чего в подавляющем большинстве случаев не делают.
Нарушения в системе ГГГ сохраняются в течение многих месяцев после ААС, и они могут играть патогенетическую роль в возникновении его рецидивов (Rasmussen et al., 2000), обеспечивая мотивацию к поиску спиртного. Причинно-следственная связь между этими явлениями прослеживается на уровне проопиомеланокортина, концентрацию которого алкоголь снижает. Этот прогормон является общим предшественником для образования b-эндорфина и адренокортикотропина, роль которых в возникновении аддиктивного поведения была продемонстрирована во многих исследованиях.
При ежедневном приеме более 72 г этанола в сутки у мужчин-алкоголиков происходит постепенная феминизация, связанная с ускорением ароматизации тестостерона в печени и образованием эстрогенов в ней (Purohit, 2000). Гипоандрогенизм, гинекомастия, атрофия яичек встречаются у 33–50 % алкоголиков. Концентрация тестостерона в плазме крови резко снижается в начале абстинентного периода и восстанавливается к исходу 10-х суток. При этом уровень тестостерон-связывающего глобулина в крови больных резко увеличивается и возвращается к варианту нормы только к 12-й неделе. У лиц с исходно высокой концентрацией тестостерона в плазме крови в меньшей степени выражены астено-невротические симптомы (апатия, нерешительность, раздражительность, тревога) во время ААС, по сравнению с пациентами, у которых уровень тестостерона был исходно низким среди больных алкоголизмом. Обычно это алкоголики II типа (Ruusa, 1996), однако у них в большей степени выражено агрессивное поведение (Staleinheim, 1998).
У женщин, употребляющих даже умеренные количества спиртных напитков, со временем возникает гиперандрогенизм за счет метаболического перехода андростендиона в тестостерон; также возникает увеличение концентрации эстрогенов из-за снижения скорости окисления эстрадиола в эстрон (Gill, 2000), обусловленное хроническим дефицитом НАД+, и снижение уровня прогестерона, особенно в предменопаузе. Считают, что увеличение базальной секреции тестостерона и андростендиона может указывать на повышенное употребление спиртного женщинами (Sakrola, 1998).
Проведение агрессивной инфузионной терапии сопровождается повышенной экскрецией физиологически активных веществ, которые способны влиять на процессы высшей нервной деятельности. Например, симптомы депрессии в постабстинентном периоде связывают с гипопролактинемией и угнетением допаминергических систем мозга во время проводимой терапии (Miller et al., 1986), особенно если в ее составе применялись нейролептики и феназепам с целью купирования клинических признаков адренергического синдрома. Рецидив «запоя» связывают с постабстинентной недостаточностью опиатергического пула и АКТГ. Часто при этом увеличивается концентрация кортизола в плазме крови, который сам по себе оказывает крайне неблагоприятное действие на функцию нейронов коры головного мозга в этих условиях (Маркова И. В. с соавт., 1999).
Даже сбалансированная инфузионная терапия может оказывать действие на функцию медиаторных и других систем организма, поскольку длительная экспозиция этанолом существенно влияет на ионный баланс организма.
Действие на обмен электролитов и КОС
Гипонатриемия, гипокалиемия, гипомагниемия и гипоцинкемия, сопутствующие алкоголизации, лежат в основе многих осложнений: нарушение ритма сердца, неврологические нарушения, демиелинизующие процессы в нервной ткани. Гипокалиемия и гипомагниемия создают электрофизиологические предпосылки для развития синдрома удлиненного Q-T. Гипоцинкемия снижает тропность ГАМК-рецепторов к ГАМК-медиатору, тем самым снижая эффективность работы одной из главных тормозных систем ЦНС. Хронический алкоголизм часто сопровождается гипонатриемией (с концентрацией натрия в плазме крови менее 134 мэкв/л). Она может быть обусловлена «адренергическим синдромом» при ААС, нарушением циркодианного ритма секреции антидиуретического гормона (Маркова И. В. с соавт., 1999), гипо-волемией (Laimis, 2000) и другими причинами, многие из которых пока не ясны.
Изменение баланса ионов натрия между матриксом и внутриклеточными секторами приводит к сдвигу экстра– и интрацеллюлярной жидкости и развитию гипер– или дегидратации. Замечено, что у лиц, потребляющих большие количества пива (более 4 л/сутки), может возникать выраженная гипонатриемия (107—98 мэкв/л), которая при отсутствии лечения завершается неврологическими нарушениями.
Острыми нарушениями КОС являются алкогольный кетоацидоз, метаболический ацидоз и алкалоз. В основе кетоацидоза лежит избыток НАДН + Н+ и блокада глюконеогенеза, во время которой энергетическими субстратами, вместо глюкозы, становятся жирные кислоты, которые окисляются только до кетокислот – бета-гидроксибутирата и ацетоацетата. Предрасполагающим фактором для развития алкогольного кетоацидоза является снижение запасов гликогена в печени (алкоголизм, многодневное интенсивное потребление спиртного, прием спиртного «без закуски», диеты с целью снижения массы тела, голодание, терапия парацетамолом, амфетаминами и т. д.).
Алкогольный кетоацидоз часто осложняется инфекцией, делирием, панкреатитом и имеет склонность к рецидивам, которые чаще всего возникают в течение 12–18 ч после его купирования в 25 % случаев.
Важно отметить, что вследствие поискового поведения больные с зависимостью к этанолу могут употреблять токсические спирты, вследствие чего к алкогольному кетоацидозу присоединяется ацидоз метаболический. Метаболический субкомпенсированный ацидоз часто возникает при голодании. К декомпенсации ацидоза приводят судороги во время ААС. Алкалоз возникает при одышке, сопутствующей «адренергическому синдрому» и рвоте.
Действие на почки
В почках, так же как и в печени, нарушаются глюконеогенез, поляризация клеточных мембран, что изменяет функцию канальцевых насосов и транспорт веществ через мембраны. Развиваются гломерулонефриты, пиелонефриты, пиелиты. Часто поражения печени и почек являются сочетанными. При ААС могут иметь место гематурия, протенурия, цилиндроурия. Эти симптомы резко усиливаются во время терапии тетурамом. Считают, что они обусловлены избыточным количеством ацетоальдегида (Станевская Т. И., 1990).
Мутагенное, канцерогенное и эмбриотоксическое действия
Указанные в заголовке действия проявляются хромосомными абберациями, увеличением процента онкологических заболеваний (особенно печени, органов пищеварения, молочных желез) и алкогольным синдромом плода. Его частота составляет от 1:300 до 1:2000 (в различных странах) или 1:3 у матерей-алкоголичек. Перинатальная смертность достигает 17 %. Дефекты умственного развития среди выживших новорожденных встречаются в 44 % (Маркова И. В., 1999). Большое значение имеет количество употребляемого беременной женщиной спиртного (см. табл. 1).
Таблица 1
Дисморфогенез и потребление этанола во время беременности

* условный стандартизованный прием равен 13,6 г 96 %-ного этанола
В плазме крови женщин, регулярно употребляющих небольшие количества алкогольных напитков, увеличен уровень тестостерона. Этанол аккумулируется в амниотической жидкости, нарушает фето-плацентарный кровоток и повреждает саму плаценту. Продукты биотрансформации этанола изменяют взаимодействие между трофо– и эмбриобластом, возникают тканевая метаплазия, «порочные» эмбриональные зачатки и дисморфогенез (см. рис. 5).

Рис. 5. «Лестница» эмбриотоксических эффектов этанола при различных типах его употребления во время беременности (по: Афанасьев В. В., 1988)
Клинические признаки алкогольного синдрома плода включают (Маркова И. В. с соавт., 1999):
задержку перинатального и постнатального роста, недоношенную беременность;
– лицевой дисморфогенез (микроцефалию, короткие и узкие глазные щели, удвоение кожной складки внутреннего угла глаза, гипоплазию верхней челюсти, малый размер челюстей, «заячью губу», складчатость ладоней);
– аномалии ЦНС (в том числе нарушение умственного развития), риск развития интракраниальных гематом, нарушение терморегуляции за счет избыточной перспирации воды через кожный по кров;
– нарушение развития суставов: шейного отдела позвоночника (слияние тел позвонков), луче-локтевых суставов (гипоплазия головки лучевой кости, лучевой синостоз), клиндактилию, аномалии строения грудной клетки, нарушение иммунного и метаболического статуса ребенка (риск инфекционных осложнений у таких детей увеличен в 4 раза);
– нарушение других функциональных систем (недоразвитие сурфактантной системы легких и возникновение синдрома дыхательных расстройств, эпизоды апноэ по центральному и периферическому типам, большая частота развития кишечных токсикозов и т. д.).
Наличие алкоголизма у одного из родителей увеличивает риск развития этого заболевания у потомства в 4–6 раз. При определенных формах алкоголизма отцов вероятность возникновения его у сыновей составляет 88 % (Арзуманов Ю. Л., с соавт., 2000).
Биологические маркеры зависимости от этанола
Патохимические реакции, происходящие в организме человека при постоянном приеме этилового алкоголя, изменяют биохимический состав и функциональную активность тканей, в результате чего могут возникать определенные химические вещества, которые изменяют функцию органа и характерны только для действия этилового алкоголя.
Биологические маркеры алкоголизма представляют собой биохимические, нейрохимические, электрофизиологические, клинические и другие показатели деятельности функциональных систем организма, которые возникают при длительном употреблении этилового алкоголя и сохраняются в организме после полного прекращения его употребления. В результате хромосомных аберраций некоторые маркеры закрепляются настолько прочно, что входят в «генетическую память» и передаются по наследству прямым родственникам. Определение биохимических и электрофизиологических (преимущественно) показателей, характеризующих зависимость от алкоголя, имеет большое значение как для клинической практики, так и для судебно-медицинских исследований.
Маркеры алкоголизма должны отвечать следующим требованиям:
– быть определяемыми в популяции алкоголиков, в том числе тех, кто находится в периоде абстиненции, и отсутствовать у здоровых людей;
– должны определяться у лиц, представляющих собой группы риска в плане возможного развития алкоголизма (лица с отягощенным наследственным анамнезом, не употребляющие спиртное);
– должны быть стабильными во времени и не изменяться при проведении детоксикации, хирургических вмешательств и других стресс-генерирующих воздействий;
– другие заболевания не должны оказывать действия на колебания значений маркера;
– должны быть стандартизованы и легко воспроизводимы.
Многие предложенные маркеры зачастую не отвечают перечисленным требованиям, однако некоторые из них могут быть приняты во внимание при проведении сложных экспертиз (см. табл. 2).
Таблица 2
Некоторые маркеры алкоголизма




Патогенез психических и соматических нарушений, возникающих в организме человека при хронической интоксикации алкоголем
Этанол прямо или опосредованно действует на субстанции внеклеточного матрикса и на рецепторы различных функциональных классов, среди которых выделяются: аллостерические ферменты (например, гексокиназный комплекс), ионные каналы (например, хлорный канал ГАМКергических систем; кальциевый канал НМДАергических систем), вторичные посредники передачи сигнала и рецептор-связывающие белки (например, ингибирующие белки М-холинореактивных систем), интегральные переносчики ионов водорода (НАД+ и НАДФ+) и т. д. (см. рис. 6).

Рис. 6. Изменения в клетках и во внеклеточном пространстве, возникающие при алкоголизации Условные обозначения:1 – нормальное состояние внеклеточного матрикса обеспечивает межклеточные контакты; 1а – нарушение структуры коллагенов, адгезивных белков и протеогликанов приводит к нарушению межклеточных контактов; сохранный билипидный слой (2) и увеличенные интермембранные пространства (2а); рецепторная передача в здоровых мембранах (3) и интернализация рецепторов, наступающая под воздействием этанола (3а); 4 и 4а – ферментативные реакции и их нарушение; 5 и 5а – лиганды и вольтаж-контролируемые ионные каналы, снижение активности одних (например, ГАМК) и увеличение активности других (например, НМДА); 6 – токсические метаболиты этанола (по: Афанасьев А. В., 1999).
Действие этанола сопровождается накоплением продуктов его биодеградации и увеличением степени эндогенной интоксикации плазмы крови (Афанасьев В. В., 1994; Афанасьев А. В., 1998). Обмен веществ в клетках изменяется, происходят неблагоприятные сдвиги во многих (если не во всех) органах и системах.
В ответ на это перестраивается нейрогуморально-гормональная регуляция функциональных систем. Этот процесс является универсальным при экспозиции «химическим фактором малой интенсивности» (Люблина Г. Н., 1974). В организме человека он осуществляется одинаковыми материальными агентами, среди которых в наибольшей степени изучена роль гормонов, медиаторов, эйкозаноидов и интерлейкинов. Эти вещества, под общим названием «аутокоиды», регулируют постоянство внутренней среды путем изменения химических синтезов в широком спектре клеток-мишений и органов-эффекторов. Возникает своеобразный адаптационный синдром, который развивается в ответ на постоянное присутствие этилового алкоголя в плазме крови (а возможно, на присутствие антител к его метаболитам), с поправкой на интенсивность и длительность действия возмущающего фактора.
Адаптация к действию этанола позволяет субъекту выжить, на какое-то время стабилизироваться и удовлетворительно сохранять свои социальные и физиологические функции. Аутокоиды можно сравнить с лекарственными веществами, у которых помимо главного действия имеются побочные эффекты. Многие из аутокоидов, первоначально являясь факторами защиты, становятся факторами агрессии и оказывают повреждающее клетки действие, которое дополняет патохимические нарушения, вызываемые этанолом и его метаболитами. Если употребление спиртного продолжается, то возникшие изменения в организме могут превысить пределы его компенсаторных возможностей и тогда начинается прогредиентный сбой в работе функциональных систем, который приводит к полиорганной недостаточности. Прекращение приема спиртного вызывает алкогольный абстинентный синдром.
«Абстинентный синдром – комплекс психопатологических, вегетативных, неврологических и соматических расстройств, проявляющихся вслед за прекращением регулярного употребления психоактивного вещества, причем постоянными симптомами являются психический и физический дискомфорт и выраженное влечение к принимаемому веществу. Абстинентный синдром при различных формах токсикомании имеет как общие неспецифичекие проявления, так и специфические черты. К неспецифическим проявлениям можно отнести резкое усиление тяги к употребляемому веществу, быстро нарастающую астенизацию, тревогу, депрессивный эффект, вегетативные нарушения (потливость, тахикардию, тошноту, гиперсаливацию, тремор и др.). Характер специфических расстройств определяется фармакологическими особенностями употребляемого вещества» (цит. по: Софронов А. Г., 2001).
Лишение «возмущающего» фактора, которым был этиловый алкоголь, сопровождается новыми адаптивными сдвигами в организме и, в первую очередь, в его нервной системе, только теперь эти сдвиги развиваются в ответ на отмену этанола. Возникает «поисковое» поведение и побудительные мотивы, заставляющие принимать спиртное. Они могут приводить к такому стилю жизни, при котором дальнейшая драма будет состоять из чередования фаз «интоксикация – абстиненция».
В представленной упрощенной модели каждому адаптивному сдвигу соответствует своя клиническая картина с последовательным развитием динамики от синдрома измененной реактивности до синдрома физической зависимости. Скорость формирования последних «во времени определяет динамику болезни» (Аронович О. М., 1978). ААС – свидетельство развернутой алкогольной болезни с характерной триадой признаков (привыканием, физической зависимостью от этанола и толерантностью к его действию). Нейроадаптивные сдвиги, возникшие в организме в ответ на постоянное введение алкоголя, гомеостазируются и впоследствии могут давать о себе знать спустя месяцы после прекращения приема алкоголя. Наиболее выраженные психовегетативные изменения в абстинентном периоде регистрируют в ближайшую неделю после отмены этанола. Известно, что стрессогенные воздействия, возникающие у лиц, находящихся в состоянии абстиненции, могут провоцировать жизнеопасные состояния. Подтверждением тому является клиническое течение инфекций, сочетанной травмы, постоперационного периода у алкоголиков, осложняющееся интоксикационным психозом, нарушениями ритма сердца, внезапно возникающей гипогликемией, нарушениями реологических свойств крови и другими состояниями. Есть наблюдения, свидетельствующие о том, что 50–60 % больных, поступающих в отделения ОРИТ с травмой, представляют собой группы риска в плане развития симптомов и осложнений ААС во время пребывания в стационаре (Spies, 1996). У «запойных» алкоголиков снижение концентрации этилового спирта в плазме крови ниже его гомеостазирующего уровня и может провоцировать клинические проявления ААС.
Наиболее сложным является вопрос о формировании аддиктивного поведения при хронической зависимости от этанола. Оно является главным фактором, обусловливающим прием спиртного, рецидив «запоя» и сокращение интервалов времени межабстинентного периода при лечении больных. Существующие модели аддиктивного поведения построены на основании данных психологических, психофизиологических, нейрохимических (и других) исследований. Результаты нейрохимических исследований имеют прямое отношение к данному оперативному руководству. Основное внимание в них уделено медиаторным сдвигам, возникающим в ЦНС. Их клинико-лабораторная оценка позволяет ориентироваться в оказании оперативной (неотложной) помощи при ААС и в проведении поддерживающей терапии в течении «спокойного» периода абстинентного состояния.
Работа медиаторных систем подчиняется определенным закономерностям. В них происходят синтез, высвобождение медиатора в синаптическую щель, его взаимодействие с постсинаптическими мембранами нейронов и т. д. Выделение этих положений дает возможность фармакологическим путем ослабить влияние системы в целом при ее избыточной активности (например, назначением глютаматблокаторов, адреноблокаторов, дофаминоблокаторов) или усилить ее воздействие при дефиците функции (например, назначением глициномиметиков, ГАМК-миметиков, холиномиметиков). Такой подход широко применяется в лечебной практике многих дисциплин: в хирургии (Базаревич Г. Я., 1982), токсикологии (Афанасьев В. В., 1994), неотложной педиатрии (Цыбулькин Э. К., 2001).
Каждая регулирующая система внутренних факторов оказывает действие на периферические функции (Аничков С. В., 1983). Изменения многих из них известны: холин– и адренергические системы контролируют большинство процессов на перферии (тахи/брадикардию; гипер/гипотензию; возбуждение/угнетение ЦНС, моторику кишечника, свертывающие свойства крови, иммунопоэз и т. д.). На первый взгляд, казалось бы, все просто: определить клинические эквиваленты медиаторного сдвига (например, по медиаторному токсикосиндрому) и назначить соответствующий ему корригирующий препарат. Такой подход частично позволяет определить степень тяжести ААС и купировать неотложные состояния, возникающие при нем, однако он не дает возможности прогнозировать осложнения, которые могут возникать в ближайший и отдаленный периоды его развития.