Глава 1. ДЕРМАТОМИОЗИТ
1.1. ОПРЕДЕЛЕНИЕ И РАСПРОСТРАНЕННОСТЬ ДЕРМАТОМИОЗИТА
Дерматомиозит (ДМ) – системное заболевание соединительной ткани, характеризующееся прогрессирующим воспалительным поражением скелетной и гладкой мускулатуры с нарушением ее двигательной функций, типичными изменениями кожи в виде эритемы и отека, а также системными проявлениями. У 25 – 30 % больных кожный синдром отсутствует. В этом случае используется термин «полимиозит» (ПМ). Реже используется термин «дерматополимиозит» или название болезни по фамилиям описавших ее авторов – болезнь Вагнера, болезнь Вагнера – Унферрихта – Хеппа. Согласно современной Международной классификации ДМ относится к группе системных заболеваний соединительной ткани и является характерным представителем аутоиммунных воспалительных миопатий.
Классификация воспалительных миопатий (Гусева Н. Г., 1994).
1. Идиопатические воспалительные миопатии:
• первичный полимиозит;
• первичный дерматомиозит;
• ювенильный дерматомиозит;
• миозит, ассоциирующийся с ДБСТ;
• миозит, ассоциирующийся с опухолями;
• миозит с включениями;
• миозит, ассоциирующийся с эозинофилией;
• оссифицирующий миозит;
• локализованный, или очаговый, миозит;
• гигантоклеточный миозит;
2. Миопатии, вызываемые инфекциями.
3. Миопатии, вызываемые лекарственными средствами и токсинами.
Впервые ДМ (острый ПМ) описали E. Wagner в 1863 г., несколько позже – Р. Нерр и H. Unverrichtt в 1887 г. К началу XX в. были выявлены различные формы болезни. В дальнейшем наблюдениями клиницистов и морфологов было доказано наличие при ДМ висцеральной патологии, а также васкулита и своеобразного поражения соединительной ткани, позволившее отнести ДМ к группе коллагеновых заболеваний. Основываясь на тяжести течения и высокой (более 50 %) летальности при ДМ, Е. М. Тареев в 1965 г. включил его в группу так называемых злокачественных или больших коллагенозов, позже трансформированную в группу диффузных болезней соединительной ткани.
Распространенность. ДМ и ПМ довольно редкие заболевания. Ежегодно диагностируется 5 новых случаев на 1 млн населения. ДМ распространен во всех климатических и географических зонах земного шара. По частоте он стоит на третьем месте среди системных заболеваний соединительной ткани вслед за системной красной волчанкой (СКВ) и системной склеродермией (ССД). ДМ (ПМ) чаще поражает женщин; соотношение по полу среди взрослых больных (женщин и мужчин), по данным большинства авторов, составляет2:1 и более. У взрослых чаще встречается ПМ, чем ДМ (2: 1), а у детей преобладает ДМ.
Отмечено два возрастных пика заболевания: 11 – 17 и 35 – 60 лет.
1.2. ЭТИОЛОГИЯ, ПАТОГЕНЕЗ И КЛАССИФИКАЦИЯ ДЕРМАТОМИОЗИТА
Этиология ДМ выяснена недостаточно. Не исключается вирусная природа заболевания (пикорнавирусы, вирус Коксаки В2, А9), а также участие бактериальных и паразитарных (риккетсиоз, шистоматоз, трихинеллез и др.) инфекций. Наиболее вероятной представляется триггерная роль вирусного фактора, вызывающего иммунорегуляторные нарушения при наличии генетической предрасположенности к заболеванию. Нельзя исключить возможность персистенции вирусов в мышцах с развитием иммунного ответа и картины ДМ (ПМ).
Описаны случаи развития ДМ и ПМ после перенесенной краснухи, герпетической инфекции, введения вакцин и сывороток, приема лекарственных препаратов. Провоцирующими факторами могут является переохлаждение, инсоляция, избыточная физическая нагрузка и др.
Опухолевый ДМ составляет 20 – 30 % от всех случаев заболевания, особенно в возрастной группе старше 50 лет.
В последние годы нашла подтверждение и наследственная предрасположенность к развитию ДМ и ПМ. Описаны случаи заболевания у кровных родственников. Современные иммуногенетические исследования показали, что в развитии идиопатических воспалительных миопатий может участвовать множество генов, а их идентификация важна для выявления генетических факторов риска ДМ (ПМ). Полученные данные позволяют предполагать, что HLA-гены на 6-й хромосоме, а именно HLA DRBl*0301 и аллели пОА1*0501 строго ассоциируются со всеми клиническими формами идиопатических воспалительных миопатий у больных белой расы. Еще ранее выявлена ассоциация ДМ (ПМ) с В8-, В14- и DR3-антигенами в европейской популяции и их ассоциация с В7 и DRW6 у темнокожих. Антигены HLA DR4 встречаются чаще при сочетании ПМ с другими ДБСТ. Хорошо известна связь антигена В8 с развитием различных аутоиммунных заболеваний, что подтверждает участие иммунных факторов в развитии ДМ. Ряд исследователей полагают, что наличие определенных гаплотипов объясняет особенности клинических форм ДМ, его сочетания с другими болезнями соединительной ткани (например, более частого сочетания со склеродермией и редкого – с ревматоидным артритом), а также выраженность иммунного компонента и т. д. Ассоциация с HLA-B8 и DR3 наиболее выражена при ювенильном ДМ и в настоящее время рассматривается как генетический маркер заболевания.
Патогенез ДМ и ПМ характеризуется синтезом широкого спектра аутоантител, направленных против цитоплазматических белков и рибонуклеиновых кислот (РНК), принимающих участие в синтезе белка (схема 1).
Эти антитела редко выявляются при других аутоиммунных заболеваниях и рассматриваются как миозитспецифические, которые условно подразделяются на четыре группы:
1) антитела к аминоацилсинтетазам транспортной РНК. Они катализируют связывание отдельных аминокислот с соответствующей тРНК;
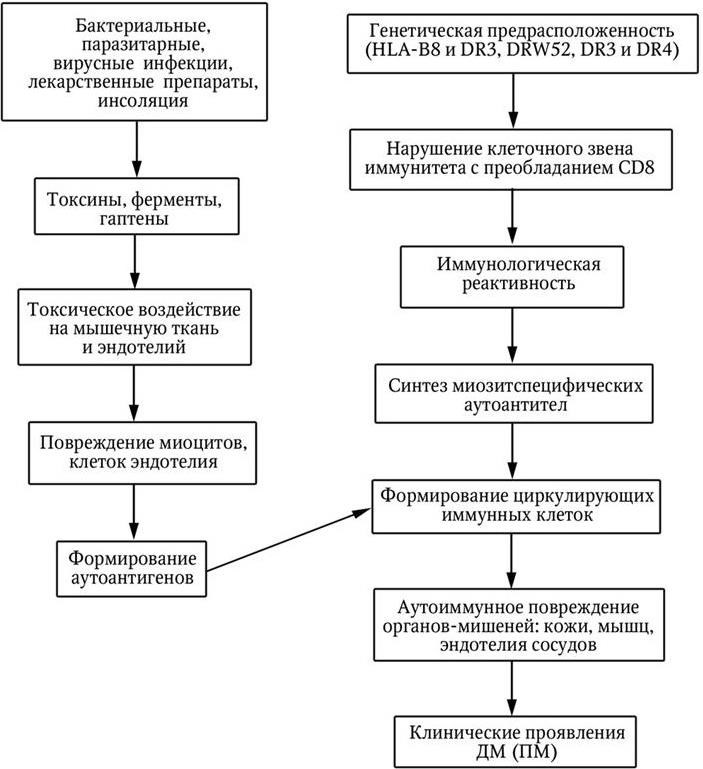
Схема 1. Патогенез ДМ (ПМ)
2) антитела, реагирующие с частицами сигнального распознавания (SRP). Эти антитела блокируют перенос вновь синтезированных белковых молекул к эндоплазматической сети;
3) антитела, реагирующие с белково-ядерным комплексом (антитела к Mi-2);
4) антитела, реагирующие с фактором элонгации i-альфа, который обеспечивает перемещение аминоацил-тРНК к рибосомам и движение вдоль полисомы.
Миозитспецифические антитела при ДМ и ПМ обнаруживаются в 40 % случаев, при этом каждый больной имеет один тип антител.
Наряду с миозитспецифическими антителами в сыворотке крови у этих больных могут присутствовать и другие типы аутоантител, неспецифичные для ДМ, включая антитела к миозину, тиреоглобулину, антитела к эндотелиальным клеткам а также ревматоидные факторы (РФ).
Одним из вероятных механизмов аутоиммунизации у больных ДМ и ПМ является перекрестная реакция между инфекционными антигенами и аутоантигенами, к которым формируются аутоантитела. Образующиеся циркулирующие иммунные комплексы (ЦИК) откладываются в тканях (мышцах, коже, сосудах и др.) и ведут к развитию иммунокомплексного воспаления. Предполагается, что аминоацил-тРНК-синтетазы могут образовывать иммуногенный комплекс с тРНК вирусного генома (например, пикорнавируса). Высказывается предположение о том, что антитела к аминоацилсинтетазам могут принимать непосредственное участие в поражении скелетных мышц. Определенное значение в развитии системных проявлений при ДМ и ПМ могут иметь антитела к эндотелиальным клеткам.
Предполагается также, что поражение мышц при ПМ связано с развитием Т-клеточной цитотоксичности против мышечных клеток, экспрессирующих антигены класса I главного комплекса гистосовместимости. Важная роль клеточного иммунитета в развитии ДМ (ПМ) подтверждается следующими данными:
1) клеточные инфильтраты в пораженных мышцах состоят преимущественно из иммунокомпетентных клеток: Т-лимфоцитов, В-лимфоцитов и макрофагов;
2) при экспозиции с мышечным антигеном лимфоциты больных ДМ (ПМ) трансформируются и увеличивают продукцию макрофагингибирующего фактора (МИФ);
3) лимфоциты больных при ДМ (ПМ) оказывают высокое цитотоксическое действие на мышечные клетки по сравнению с контрольными лимфоцитами;
4) лимфоциты больных ДМ (ПМ) высвобождают лимфотоксин, способный нарушать мышечный метаболизм, а также особый фактор, который ингибирует ионы кальция, связанные с саркоплазматическим ретикулумом и контрактильной способностью мышц;
5) лимфоциты животных с экспериментальным ПМ оказывают цитотоксическое действие на скелетные мышцы.
Кроме того, были выявлены некоторые иммуноморфологические различия между ДМ и ПМ. Так, в воспалительных инфильтратах при ДМ было обнаружено преобладание CD4+ Т-лимфоцитов, увеличение экспрессии молекул адгезии VCAM (сосудистые клеточные адгезивные молекулы) на воспалительных и эндотелиальных клетках, включая дерму. При ПМ в инфильтратах превалируют CD8+ Т-лимфоциты, экспрессирующие молекулы la/DR и другие маркеры активации. Высокая экспрессия молекул la/DR обнаружена и на мышечных волокнах. Показано, что периферические мононуклеары при ДМ оказывают повреждающее действие на фибробласты кожи в культуре ткани.
Таким образом, Т-клетки и макрофаги активно участвуют в развитии нарушений клеточных взаимодействий при поражении соединительной ткани и, следовательно, в целом ряде звеньев патогенеза ДМ (ПМ).
Исследования последних лет доказали значение цитокинов в развитии ДМ (ПМ). Экспрессия цитокинов, главным образом провоспалительных – ИЛ-1α, ИЛ-1β и ТФР-β (трансформирующий фактор роста в), выявлена в мышечной ткани при идиопатических воспалительных миопатиях (ДМ, ПМ и миозит с включениями). Преобладание экспрессии ИЛ-1α в стенках сосудов у больных ДМ (ПМ) указывает на важную роль данного цитокина в развитии патологического процесса.
Взаимодействие антигенов с аутоантителами ведет к формированию циркулирующих иммунных комплексов. Патогенетическая роль ЦИК обсуждается в связи с их взаимодействием с Fc-рецепторами лимфоцитов, обусловливающим увеличение синтеза иммуноглобулинов (индуцируют увеличение ЦИК, т. е. развивается порочный круг), и c высвобождением лимфокинов, участвующих в развитии воспаления и повреждения мышц.
Отложение иммунных комплексов в тканях (мышцах, коже, сосудах и др.) ведет к развитию иммунокомплексного воспаления. Все это свидетельствует о несомненном участии и ведущей роли иммунных нарушений в локальных и общих проявлениях ДМ (ПМ).
Развитие опухолевого ДМ связывается как с возможностью прямого токсического действия на мышцы опухолевых субстанций, так и c развитием аутоиммунной реакции вследствие общности антигенов опухолевой и мышечной ткани. Классификация. В настоящее время наиболее широко используется классификация ДМ и ПМ, предложенная A. Bohan и Y. Peter (1975).
Классификация ДМ и ПМ по A. Bohan и Y. Peter в 1975 г.
1. Первичный (идиопатический) полимиозит.
2. Первичный (идиопатический) дерматомиозит.
3. Дерматомиозит (или полимиозит) в сочетании с неоплазмой.
4. Ювенильный дерматомиозит или полимиозит в сочетании с васкулитом.
5. Дерматомиозит (или полимиозит) в сочетании с ДБСТ.
Выделяют также острое, подострое и хроническое течение ДМ и ПМ.
Острое течение характеризуется лихорадкой, генерализованным поражением поперечнополосатой мускулатуры, вплоть до полной обездвиженности, прогрессирующей дисфагией, висцеритами, эритематозным поражением кожи, развивающимися в течение первых 6 мес. от начала заболевания.
Подострое течение характеризуется более медленным нарастанием симптомов заболевания, но через 1 – 2 года после появления первых клинических признаков наблюдается развернутая картина ДМ (ПМ) с тяжелыми поражениями мышц, кожи и висцеральными проявлениями.
Хроническое течение характеризуется медленным развертыванием клинических проявлений заболевания в течение нескольких лет, преобладанием процессов атрофии и склероза мышечной ткани, поражением кожных покровов в виде гиперпигментации, гиперкератоза, редкими висцеральными проявлениями.
По степени клинико-лабораторной активности выделяют низкую (1), среднюю (2), высокую (3) активность и ремиссию.
1.3. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ДЕРМАТОМИОЗИТА
Клинические проявления. Начало заболевания может быть острым с лихорадкой до 38 – 39 °С, проливными потами, похуданием. Но чаще симптоматика развивается постепенно, характеризуясь преимущественно кожными (ДМ) и мышечными проявлениями.
Поражение кожи при ДМ полиморфно: преобладают эритема, отек и дерматит (см. цв. вкл., рис. 1.1) преимущественно на открытых участках тела. Наблюдаются папулезные, буллезные, петехиальные высыпания, телеангиэктазии, очаги пигментации и депигментации, гиперкератоза.
Характерны периорбитальный отек и эритема, имеющая своеобразный лиловый оттенок (симптом «очков»), играющий важную роль при постановке диагноза (см. цв. вкл., рис. 1.2).
Яркая эритема чаще локализуется на лице, шее, в зоне декольте, над проксимальными межфаланговыми и пястно-фаланговыми суставами (синдром Готтрона) (см. цв. вкл., рис. 1.3), на наружной поверхности предплечья и плеча, передней поверхности бедер и голеней.
Отек лица и конечностей преимущественно над пораженными мышцами имеет тестоватый характер. Нередко наблюдаются трофические нарушения в виде сухости кожи, ломкости ногтей, выпадения волос и др. При хроническом течении ДМ развиваются кожные изменения по типу пойкилодермии, имеются очаги пигментации и депигментации, множественные телеангиэктазии, истончение кожи, сухость, участки гиперкератоза. У половины больных ДМ отмечаются одновременно конъюнктивит, стоматит, отеки зева голосовых связок.
Таким образом, характерные изменения кожи и слизистых позволяют заподозрить ДМ уже при первом взгляде на больного.
Поражение скелетных мышц является ведущим признаком ДМ и ПМ. Характерно развитие тяжелого, нередко некротического миозита с преимущественным поражением мышц проксимальных отделов конечностей, плечевого и тазового пояса, шеи, спины, глотки, верхних отделов пищевода, сфинктеров.
Клинически отмечают боль в мышцах, плотность или тестоватый характер пораженных мышц, увеличение их в объеме, болезненность при пальпации. Доминирующим признаком служит неуклонно прогрессирующая мышечная слабость, выражающаяся в значительном ограничении активных движений больных, что не позволяет им самостоятельно встать, сесть, поднять ногу на ступеньку (симптом «автобуса»), удержать какой-либо предмет в руке, причесаться, одеться (симптом «рубашки»). При поражении мышц шеи и спины больные не могут самостоятельно приподнять голову с подушки или удержать ее в положении сидя (голова падает на грудь), не могут самостоятельно сесть и приподняться с постели. Практически затруднены все движения, связанные с участием проксимальных мышц конечностей (плечевой и тазовый пояс), в то время как в дистальных отделах конечностей (в кистях и стопах) сохраняются удовлетворительная сила и полный объем движений.
Постепенное вовлечение в процесс мышц шеи и спины усугубляет тяжесть состояния больных, которые в связи с нарастающей инвалидизацией и обездвиженностью требуют постоянного ухода.
Вовлечение в процесс глоточных мышц вызывают дисфагию (поперхивание при глотании), возможна аспирация пищи в трахею. В отличие от дисфагии, наблюдающейся при системной склеродермии, у больных ДМ затруднено глотание как твердой, так и жидкой пищи, которая иногда выливается через нос. Поражаются преимущественно верхние отделы пищевода, мышцы мягкого нёба, языка; развивающаяся псевдобульбарная симптоматика имитирует неврологическое заболевание.
Поражение межреберных мышц и диафрагмы, ведущее к ограничению подвижности и снижению жизненной емкости легких, способствует развитию пневмонических осложнений – одной из основных причин летального исхода при ДМ.
При поражении мышц гортани появляются носовой оттенок голоса (дисфония), охриплость, вплоть до афонии. Поражение мышц сфинктеров ведет к расстройству их деятельности.
Скорость развития симптоматики зависит в основном от характера течения болезни. При острой форме заболевания тяжелая мышечная слабость может появиться в течение первых 2 – 3 недель, нередко сочетается при этом с миоглобинурией. Чаще симптомы ПМ развиваются постепенно – в течение 3 – 6 мес. (подострое течение). Мышечная слабость при хроническом ДМ (ПМ) может нарастать в течение ряда лет. Всегда сохраняется характерная локализация процесса – проксимальные отделы мышц конечностей.
Мышцы лица поражаются редко, вовлечение в процесс глазных мышц при ДМ (ПМ) практически не наблюдается. Однако Н. Г. Гусева (2004) относит к варианту ДМ (ПМ) также и регионарную форму – сегментарный полимиозит с поражением отдельных групп мышц (плечевых, лопаточных, бедренных) склерозирующего или воспалительного характера. Сюда же они включают орбитальный миозит, при котором наблюдаются птоз, диплопия, а также ряд других локальных миозитов. Для ДМ характерно системное поражение мышц, в тяжелых случаях заболевания достигающее степени «мышечной чахотки» (миофтиза).
С целью стандартизации оценки состояния больного по ведущему признаку заболевания – мышечной слабости, предлагается градация степени ее выраженности (по Н. Г. Гусевой).
Градация степени и выраженности мышечной слабости при ДМ (ПМ):
I. Нет нарушений в момент осмотра.
II. Нет нарушений при осмотре, но имеются небольшая слабость и снижение толерантности к физической нагрузке.
III. Небольшая атрофия одной или более мышечных групп без функциональных нарушений.
IV. Нарушения функций: неспособность бегать, при сохранении возможности подниматься и спускаться по лестнице без опоры и помощи рук.
V. Выраженная мышечная слабость, лордоз; больной неспособен идти по лестнице (спускаться) или подниматься со стула без помощи рук (или посторонней помощи).
VI. Больной не способен встать без посторонней помощи.
Степени I – III характеризуются минимальной выраженностью или отсутствием мышечной слабости, IV степень – средней ее выраженностью, а приVиVI степени наблюдаются тяжелые инвалидизирующие функциональные нарушения. Предлагаемая градация нарушений мышечных функций при ДМ (ПМ) применима для сравнительной характеристики групп больных, оценки динамики процесса и, следовательно, мониторинга терапии. В целях стандартизации и объективизации мышечной патологии, контроля эффективности лечения иногда используют и различные биомеханические тесты, разработанные для верхних и нижних конечностей.
Следует подчеркнуть, что миалгии не превалируют в картине болезни ДМ (ПМ) и не столь выражены, как при ревматической полимиалгии (РПМ), где они определяют наблюдающиеся ограничения движений. Для ДМ (ПМ) характерна псевдопаралитическая слабость мышц, что наряду с псевдобульбарным синдромом нередко заставляет врача ошибочно предполагать у больного наличие неврологической патологии.
Тяжесть состояния и инвалидизации больных ДМ (ПМ) обусловлена также нередким последующим развитием сухожильно-мышечных контрактур, атрофией и кальцинозом ранее пораженных групп мышц. Кальцинируются обычно участки фасции, подкожной клетчатки, прилежащие к пораженным мышцам, т. е. преимущественно в области плечевого и тазового пояса. Кальциноз чаще встречается при ДМ (ПМ) у детей, но может также осложнять течение ДМ (ПМ) взрослых, особенно при отсутствии адекватной и своевременной терапии кортикостероидами. При ювенильном ДМ (ПМ) кальциноз развивается примерно через 16 мес. после начала заболевания. Массивные участки кальциноза могут быть резко болезненны, а их распространение на периартикулярные ткани приводит к обездвиженности больных. Расположенные под кожей кальцинаты частично отторгаются в виде крошковатых масс, приводя к изъязвлениям, а иногда и нагноениям.
Суставной синдром не является ведущим в клинике ДМ и ПМ и выявляется у 27,7 % взрослых пациентов (Насонов Е. Л., 2005). Чаще поражаются локтевые, плечевые, коленные суставы и кисти рук. Характерны артралгии, поражения периартикулярных тканей; артриты возникают редко. Нарушение функции суставов и контрактуры чаще связаны с поражением мышц.
Синдром Рейно (СР) отмечается у 20 % больных ДМ (ПМ), чаще у детей. Он наиболее типичен для сочетанных форм со склеродермией. При идиопатическом ДМ синдром Рейно в основном имеет двухфазный характер, который не приводит к образованию трофических язв. При капилляроскопии выявляют нарушения микроциркуляторного русла, сочетающиеся как с синдромом Рейно, так и с васкулитами: расширение капиллярных петель, замедление кровотока и сладж-синдром, аваскулярные поля. Эти изменения более характерны для ДМ, чем для ПМ (Гусева Н. Г., 2004). Они не имеют четкой корреляции с выраженностью и активностью миозита, хотя и уменьшаются при длительной ремиссии; чаще обнаруживаются у больных с синдромом Рейно, поражением кожи, суставов и легких, а также при overlаp-синдроме.
Поражение внутренних органов обычно встречается у большинства больных ДМ (ПМ), но не превалирует в картине болезни, как, например, при ССД и СКВ.
Поражение сердца, особенно миокарда, нередко наблюдается у больных ДМ (ПМ) и может стать причиной смерти. Клинические, функциональные и морфологические сопоставления показали относительную скудность клинической симптоматики и важную роль инструментальных методов в выявлении данной патологии. В большинстве случаев поражение сердца развивается при остром варианте течения ДМ (ПМ) и характеризуется тахикардией, умеренным расширением границ сердца, приглушением тонов чаще в области верхушки, аритмиями, гипотонией. Эти признаки свидетельствуют о наличии застойной сердечной недостаточности, связанной с развитием миокардита или миокардиофиброза.
В наблюдениях Е. Л. Насонова (2005) изменения миокарда отмечены у 71 % больных ДМ (ПМ), но лишь в 32,3 % случаев они были связаны с основным заболеванием, в 22,3 % – ссопутствующими (атеросклероз, гипертоническая болезнь), а в 16,9 % случаев были обусловлены приемом больших доз кортикостероидов (стероидная миокардиопатия). При электрокардиографическом исследовании выявляются наиболее характерные нарушения ритма и проводимости – блокады различных степеней, изменения зубца Т и смещение интервала S – Т. Желудочковая экстрасистолия, фибрилляция предсердий, бигеминия иногда могут наблюдаться в различное время у одного и того же больного, нередко ассоциируясь с внутрижелудочковым нарушением проводимости – блокадами левой или правой ножки предсердно-желудочкового пучка и др. Такие нарушения ритма, как предсердная и суправентрикулярная пароксизмальная тахикардия бигеминия, могут отсутствовать при обычном электрокардиографическом исследовании, но обнаруживаться при 24-часовом холтеровском мониторировании.
Таким образом, поражение миокарда при ДМ (ПМ) может протекать в виде выраженного кардита, но чаще развиваются субклинические варианты, что диктует необходимость использования современных методов функционального исследования сердца для выявления и уточнения характера его поражения.
Новые неинвазивные методы исследования сердца позволили подтвердить частоту и различный характер его поражения при ДМ (ПМ). Так, при использовании данных эхокардиографии (ЭхоКГ), суточного мониторирования, перфузионной сцинтиграфии с 201Т1 и исследовании центральной гемодинамики были выявлены изменения со стороны сердца у всех обследованных больных. Одновременно у них отмечался и высокий уровень МВ фракции креатинкиназы в крови.
Как показали наблюдения Н. Г. Гусевой (2004), прижизненная биопсия и патологоанатомические исследования в миокарде больных ДМ (ПМ) обнаруживают изменения, в значительной степени сходные с таковыми в скелетных мышцах – мононуклеарную инфильтрацию, иногда некроз и атрофию мышечных волокон. Генез этих изменений при ДМ (ПМ) объясняется наличием миокардита, но, возможно, обусловлен и ишемическими изменениями в связи с поражением мелких сосудов коронарного русла. Для обозначения этой патологии иногда используется термин «полимиозитная кардиопатия».
Описаны случаи дилатационной кардиомиопатии. При высокой клинико-лабораторной активности может наблюдаться развитие констриктивного перикардита. Поражение легких у больных ДМ обусловлено рядом факторов, среди которых выделяют мышечный синдром (гиповентиляция), присутствие инфекционных агентов, аспирацию при нарушении глотания, развитие интерстициальной пневмонии и фиброзирующего альвеолита.
Мышечная слабость, распространяющаяся на дыхательные мышцы, включая диафрагму, может быть причиной снижения вентиляционной функции легких. Клинически отмечается частое и поверхностное дыхание, инспираторная одышка, развивается гипостатическая пневмония. Дисфагия с аспирацией жидкости и пищи в легкие обусловливает развитие аспирационной пневмонии. Поражение легких выявляется у 5 – 46 % больных ДМ (ПМ), главным образом в виде интерстициальной пневмонии, фиброзирующего альвеолита и фиброза. Диагностируется на основании клинико-рентгенологических, функциональных и морфологических (биопсия) данных.
Одышка и кашель, хрипы и крепитация наблюдаются при выраженном поражении легких. Легочные функциональные тесты указывают на преимущественно рестриктивный тип нарушений со снижением общей и жизненной емкости легких; гипоксемия характеризуется умеренным снижением диффузионной способности легких.
Патология легких при ДМ (ПМ) имеет сложный генез и варьирует в широких пределах. Выделяют определенные субтипы интерстициального поражения легких, которые следует учитывать при диагностике и лечении ДМ (ПМ):
I. Острый или подострый тип, который характеризуется тяжелой быстропрогрессирующей одышкой и нарастающей гипоксемией уже в первые месяцы заболевания, аналогично синдрому Хаммена – Рича.
II. Хронический тип с медленнопрогрессирующей одышкой.
III. Асимптомный тип, который протекает субклинически, выявляется при рентгенологическом и функциональном исследовании легких.
Первый тип интерстициального поражения легких имеет наихудший прогноз и требует ранней активной терапии (глюкокортикостероидами (ГКС), цитостатиками и др.).
Полимиозит может предшествовать легочной патологии, развиться после поражения легких или возникать одновременно с легочными проявлениями. Выделяется также и «амиопатический» ДМ, который может ассоциироваться с тяжелым быстропрогрессирующим поражением легких.
Легочный фиброз, обусловленный интерстициальным поражением ткани легких, легочным васкулитом и развитием септально-альвеолярного склероза, отмечается у 5 – 10 % больных. Он характеризуется нарастающей инспираторной одышкой, сухим кашлем, крепитирующими хрипами в нижних отделах легких, нарастающей дыхательной недостаточностью.
Необходимо иметь в виду возможность развития опухолевого, чаще метастатического, процесса в легких.
Поражения желудочно-кишечного тракта (ЖКТ) отмечаются нередко и проявляются нарастающей дисфагией, отсутствием аппетита, иногда – болью в животе и гастроэнтероколитом.
Ведущей симптоматикой поражения желудочно-кишечного тракта при ДМ (ПМ) является дисфагия. Дисфагия развивается вследствие снижения контрактильной силы фарингеальных мышц и мышц верхнего отдела пищевода, нарушения перистальтики, слабости мышц мягкого нёба и языка. Это обусловливает поперхивание, нарушение глотания твердой и жидкой пищи, которая может выливаться через нос. Фарингеально-пищеводная дисфагия – важный дифференциально-диагностический признак ДМ (ПМ). Голос приобретает носовой оттенок. Дисфония нередко сочетается с дисфагией и у тяжелобольных иногда переходит в афонию.
В отличие от ССД при ДМ (ПМ) поражаются верхние отделы пищевода и глоточное кольцо, поэтому клиническая и рентгенологическая картины различны. При склеродермии жидкая пища проходит хорошо, не выливается через нос, однако рентгенологические признаки поражения и осложнения склеродермического эзофагита выражены сильнее, чем у больных с ДМ (ПМ).
Тяжелая прогрессирующая дисфагия, когда твердая пища срыгивается, а жидкая выливается через нос, из-за возможности аспирации представляет непосредственную угрозу жизни больного и является прямым показанием к срочной терапии максимальными дозами кортикостероидов.
При вовлечении в процесс пищеводного сфинктера возможно развитие рефлюкс-эзофагита.
Описаны отдельные случаи ДМ (ПМ) с желудочно-кишечными кровотечениями, перфорацией желудка, в основе которых лежат васкулит и некрозы, возникающие в пищеварительном тракте.
Умеренное увеличение печени с изменением функциональных проб наблюдается приблизительно у 1/3 больных; реже выявляется гепатолиенальный и железисто-селезеночный синдромы.
Поражения почек при ДМ (ПМ) встречаются относительно редко. При остром течении тяжелая персистирующая миоглобинурия может привести к развитию почечной недостаточности. Среди больных ДМ (ПМ) у 41,5 % отмечается транзиторная протеинурия с микрогематурией и цилиндрурией (Мазуров В. И., Беляева И. Б., 2005). При остром течении ДМ (ПМ) у отдельных больных могут наблюдаться развитие гломерулонефрита, сосудистая патология почек с фибриноидными изменениями артериол и тромбозом.
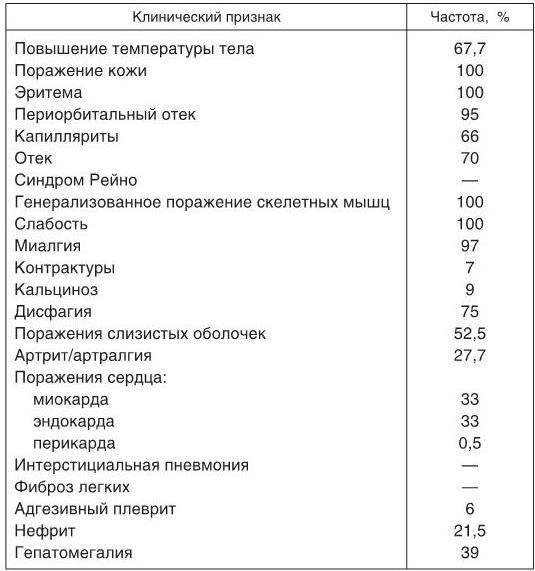
Таблица 1
Частота основных клинических проявлений ДМ (ПМ)
Эндокринные нарушения проявляются изменениями функциональной активности половых желез, гипофизарно-надпочечниковой системы, которые могут быть связаны как с тяжестью заболевания и васкулитом, так и с проводимой стероидной терапией.
Поражение нервной системы встречается редко и характеризуется развитием псевдоневрологической симптоматики, хотя у отдельных больных возможно развитие слабо выраженного полиневрита и даже поражений центральной нервной системы (ЦНС) за счет васкулита. Наиболее часто отмечаются вегетативные расстройства. Изредка наблюдаются нарушения психики, эмоциональная неустойчивость больных, которые могут быть связаны с приемом высоких доз ГКС. Частота основных клинических проявлений ДМ (ПМ) по данным Н. Г. Гусевой (2004) представлена в табл. 1.
1.4. КЛИНИКО-ИММУНОЛОГИЧЕСКИЕ ПОДТИПЫ ДЕРМАТОМИОЗИТА
Кроме классических вариантов ДМ и ПМ можно выделить несколько клинико-иммунологических подтипов, развитие которых ассоциируется с синтезом различных типов антител. Эти подтипы различаются не только по спектру клинических проявлений, но и по иммуногенетическим маркерам, прогнозу, ответу на применение ГКС.
Синтез антител к аминоацил-тРНК-синтетазам ассоциируется с развитием так называемого антисинтетазного синдрома, для которого характерны следующие основные признаки: острое начало миозита, интерстициальное поражение легких, лихорадка, симметричный артрит, синдром Рейно, «рука механика», неполный ответ на применение ГКС с частым обострением при снижении дозы препарата, начало заболевания преимущественно в весеннее время. Характерным проявлением антисинтетазного синдрома является интерстициальное поражение легких, которое выявляется у 50 – 70 % больных с наличием антител Jо-1. Развитие артрита чаще наблюдается у больных с наличием антител Jо-1 (57 – 100 %), чем у больных с другими формами миозита. Артрит, как правило, неэрозивный, характеризуется наиболее частым вовлечением в процесс мелких суставов кистей и лучезапястных суставов. Синдром Рейно при антисинтетазном синдроме наблюдается в 60 % случаев.
При воспалительных миопатиях, сопровождающихся синтезом антител SRP (неантисинтетазные цитоплазматические антитела), чаще выявляются клинические признаки ПМ. Отмечаются следующие клинические особенности: более частое поражение мужчин, чем женщин (6: 1), острое начало и тяжелое течение миозита, высокая частота поражения сердца, плохой ответ на применение ГКС.
Антитела РМ-1, относящиеся к группе антиядерных антител, наиболее часто выявляются при перекрестном синдроме ПМ – ССД. Реже эти антитела выявляются при ювенильном ДМ.
Особенностями миозита «с включениями» являются развитие слабости и атрофии не только проксимальных, но и дистальных групп мышц, умеренное или минимальное увеличение мышечных ферментов, редкая ассоциация с ДБСТ и злокачественными новообразованиями, резистентность к лечению ГКС.
Клинические особенности ПМ, ДМ и миозита с включениями представлены в табл. 2.
При миозите, ассоциирующемся с ДБСТ, в клинической картине превалирует мышечная слабость. Среди заболеваний соединительной ткани, сочетающихся с ДМ (ПМ), на первом месте стоит ССД, затем ревматоидный артрит (РА) и СКВ, реже наблюдаются сочетания с синдромом Шегрена, узелковым полиартериитом (УП), а также с саркоидозом и др.
Таблица 2
Основные клинические симптомы дерматомиозита, полимиозита и миозита с включениями (Насонов Е. Л., 2005)

Существуют три варианта сочетания ДМ (ПМ) с другими заболеваниями соединительной ткани. Первый, когда к картине ДМ (ПМ) присоединяется отдельный(ые) признак(и) другого заболевания, например СКВ или ССД. При втором варианте имеются одновременно признаки ДМ (ПМ) и другого ревматического заболевания, образуя смешанное заболевание соединительной ткани, или overlap-синдром. Третий – развитие ДМ (ПМ) на фоне других диффузных болезней соединительной ткани, например ССД или СКВ, когда ДМ (ПМ) является синдромом основного заболевания. В каждом из таких наблюдений необходимо исключать симптоматику, сходную с другими ревматическими заболеваниями. Так, кожные изменения, особенно при выраженной эритеме и трофических нарушениях, близки наблюдающимся при хронической красной волчанке, а при наличии отека и маскообразности являются склеродермоподобными. Мышечный синдром (без кожных изменений) нередко трактуется как ревматическая полимиалгия, первичная фибромиалгия, а в последнее время и как диффузный фасциит. Общая картина заболевания, наблюдение и использование диагностических критериев позволяют правильно диагностировать ДМ (ПМ).
В основе наблюдающегося полиморфизма ДМ (ПМ), особенно его сочетаний с диффузными заболеваниями соединительной ткани, лежат иммуногенетические особенности, которые проявляются «неравновесным сцеплением» и комбинацией не только определенных генов, ответственных за заболевание, но и связанных с ними клинических симптомокомплексов, формирующих overlap-синдром. Этим можно объяснить и наличие у одного больного признаков трех и даже четырех ревматических заболеваний, включая ДМ (ПМ).
Миозит, ассоциирующийся с опухолями, составляет 20 % всех случаев идиопатической воспалительной миопатии (ИВМ). Частота сочетания злокачественных опухолей с ДМ (ПМ) колеблется от 6,7 до 34 %, что в значительной мере зависит от возраста наблюдающихся больных. Хотя при ряде системных заболеваний соединительной ткани отмечаются определенные ассоциации с неоплазмой (при ССД, синдроме Шегрена и др.), ДМ (ПМ) представляет наиболее яркий и характерный пример паранеопластического процесса при ревматических болезнях. Частота возникновения опухолей при ДМ в 5 – 11 раз выше, чем в общей популяции. Однако у детей сочетание ДМ (ПМ) с опухолями наблюдается редко. В последующие возрастные периоды ассоциация ДМ с неоплазмой возрастает и достигает 40 – 50 % у больных старше 40 лет. Таким образом, развитие ДМ (ПМ) в возрасте после 40 лет может рассматриваться как маркер опухолевого процесса.
Сочетание ДМ (ПМ) с неоплазмой встречается чаще у мужчин, чем у женщин, хотя среди наблюдавшихся Н. Г. Гусевой это преобладание не столь значительно. При сочетании с опухолевым процессом превалирует картина ДМ, а не ПМ, что может быть обусловлено генерализованным характером паранеопластических реакций. При этом следует отметить, что ПМ чаще отмечается у женщин.
Как один из механизмов развития опухолевого ДМ (ПМ) обсуждается возможность перекрестных реакций между неопластическим и мышечным антигенами, активация латентной вирусной инфекции с последующим включением иммунных механизмов и развитием ДМ (ПМ), воздействие вирусного или иных факторов на иммунный аппарат с экспрессией одновременно ДМ и опухолевого роста. Антигенное воздействие опухоли и включение иммунных механизмов развития паранеопластического ДМ (ПМ), очевидно, возможны на самых ранних этапах бластогенеза, когда тщательное общеклиническое исследование не обнаруживает неоплазмы, что не является доказательством ее отсутствия.
Чаще злокачественную опухоль выявляют во время обследования больного с мышечной слабостью или уже установленным диагнозом ДМ (ПМ), при этом мышечная слабость как проявление ДМ (ПМ) может предшествовать обнаружению неоплазмы на 1 – 2 года и раньше. Реже опухоль диагностируют до появления признаков ДМ (ПМ).
Характер локализации неоплазм при ДМ (ПМ) отражает общие возрастные и популяционные закономерности, за исключением более высокой частоты овариальных карцином.
При вторичном ДМ (ПМ) чаще наблюдаются неоплазмы органов половой сферы, легких, желудочно-кишечного тракта, почек. Реже встречаются лимфомы, саркомы, лейкоз, злокачественная меланома и т. д. Связь ДМ с опухолью четко аргументируется случаями излечения больных после удаления опухоли и, наоборот, рецидивом или прогрессированием заболевания при наличии метастазов.
Клинические проявления паранеопластического ДМ (ПМ) могут быть идентичны идиопатическому (первичному) ДМ, однако отмечается торпидность к проводимой терапии (стероидорезистентность), в том числе и к относительно большим дозам кортикостероидов.
В молодом возрасте сочетание ДМ (ПМ) с опухолью встречается редко, однако и в этих случаях необходима онкологическая настороженность, особенно при торпидном к лечению и атипично протекающем ДМ (ПМ).
Особое внимание следует обратить на возможность развития у этой группы пациентов лимфопролиферативных заболеваний, которые могут иметь свои особенности течения и маскировать более яркими критическими проявлениями ДМ (ПМ).
Таким образом, к «факторам риска» выявления злокачественных опухолей, помимо возраста и пола, следует отнести атипичное течение и рефрактерность ДМ (ПМ) к проводимой терапии, и наоборот – наличие миозитспецифических антител, которые практически не наблюдаются при опухолевом ДМ и ПМ. Нормальные значения креатинфосфокиназы (КФК) у больных с типичными проявлениями миозита могут также указывать на его связь со злокачественными новообразованиями.
Детский (ювенильный) дерматомиозит встречается приблизительно с одинаковой частотой у мальчиков и девочек, но, по данным некоторых авторов, может превалировать у мальчиков.
Е. Л. Насонов (2005) выделил ДМ (ПМ) у детей как особую форму заболевания в связи с выраженностью симптомов и частотой встречаемости васкулита. Соотношение ДМ и ПМ в этой группе примерно 2: 1. Выявляется бимодальное распределение ДМ/ПМ в детском возрасте: дети от 5 до 9 лет – 3,7 случая на 1 млн в год, детей от 10 до 14 лет – 4,3 случая на 1 млн в год. При начале заболевания до 7 лет отмечается его более легкое течение.
Прогноз ДМ в детском возрасте оценивается различно. Из 118 детей с ДМ (ПМ), наблюдавшихся Л. А. Исаевой и М. А. Жвания, 13 больных умерли, у 20 развилась тяжелая инвалидность, у остальных лечение сопровождалось улучшением состояния со снижением активности, а в ряде случаев и клинико-лабораторной ремиссией. У 10 больных длительность ремиссии составила 10 – 13 лет, что позволяет условно говорить о выздоровлении. В настоящее время с учетом более активной терапии, включающей пульс-терапию кортикостероидами, применение циклофосфамида, циклоспорина А и внутривенное введение иммуноглобулина (IgG), прогноз ювенильного ДМ (ПМ) значительно улучшился.
Клинические проявления ювенильного ДМ (ПМ) в целом сходны с таковыми у взрослых, однако имеются и некоторые особенности, связанные с выраженными васкулитами и микроангиопатиями, нередко более острым началом, экссудативным компонентом (отеки), синовитами с последующим развитием распространенного кальциноза тканей.
В большинстве случаев заболевание начинается с лихорадки, резкой боли в мышцах, кистях и стопах, нарастающей мышечной и общей слабости, прогрессирующего снижения массы тела.
Поражения кожи отмечаются у большинства больных и проявляются в виде лилового оттенка лица или характерной гелиотропной эритемы в периорбитальных областях, высыпаний в области лба, век, иногда щек, шеи, передней и задней поверхности грудной клетки, конечностей. Нередко одновременно развивается отек кожи, подкожной клетчатки, периартикулярных тканей, иногда имитирующий синовиты или действительно сочетающийся с ними. В области ногтевого ложа обнаруживаются микронекрозы (васкулит) и телеангиэктазии, а над суставами кисти – эритемы Готтрона с характерным цианотично-белесоватым оттенком, атрофией и восковидным шелушением. При тяжелых формах васкулита возможны изъязвления и некрозы кожи, висцеральных органов (кишечника и др.).
Поражение мышц характеризуется нарастанием мышечной слабости и обездвиженности больных, часто более выраженным болевым компонентом, что иногда трудно дифференцировать от полиартрита. Появляющиеся дисфагия и дисфония не позволяют сомневаться в диагнозе ДМ (ПМ). Особенно неблагоприятным фактором (симптомом) является нарастающее поражение дыхательной мускулатуры с развитием дыхательной недостаточности, аспирационной и гипостатической пневмонии. При более постепенном развитии клинической картины болезни вначале появляются небольшая мышечная слабость, иногда – кожные высыпания, умеренные синовиты и тендиниты, синдром Рейно. При поражении мышц тазового пояса ребенок часто падает, затем выявляется ограничение движений, развиваются стойкие контрактуры в суставах конечностей, атрофия мышц и, наконец, выраженная, иногда генерализованная кальцинация в области подкожной клетчатки и мышц. Все это приводит к почти полной обездвиженности и тяжелой инвалидизации больных ювенильным ДМ (ПМ). Кальциевые отложения появляются в среднем через 16 мес. после начала заболевания, изъязвляются, иногда инфицируются и при преимущественном отложении в области конечностей, тазового и плечевого пояса способствуют образованию контрактур, ограничению движений и иммобилизации больных в подростковом и молодом возрасте. Показано, что кальциноз развивается у 65 % не леченных стероидами пациентов. Механизмы кальцинации мало изучены, но их связь с предшествующим воспалением, васкулопатией и некротическими изменениями не вызывает сомнений. Обсуждается роль развития щелочной реакции и увеличения содержания щелочной фосфатазы в месте воспаления, локального повышения уровня кальция, фосфора и гликозаминогликанов. При этом лабораторные параметры фосфорно-кальциевого метаболизма остаются в норме.
Таким образом, ДМ (ПМ) у детей имеет определенные отличия:
1) наличие распространенного васкулита, проявляющегося клинически и особенно при морфологическом исследовании;
2) частое развитие подкожного кальциноза (в 5 раз чаще, чем у взрослых), что характеризует активный и прогрессирующий процесс развития заболевания;
3) редкое сочетание с опухолевым процессом.
Сочетание ДМ с другими заболеваниями соединительной ткани у детей наблюдаются сравнительно редко.
В. И. Мазуров и И. Б. Беляева (2005) выделяют следующие формы хронического течения ПМ: вариант Вагнера – Унферрихта (классическое течение), псевдомиопатическую, псевдомиастеническую, псевдоамиотрофическую, миосклеротическую, миалгическую и форму с синдромом Мак-Ардля.
Псевдомиопатическая форма ПМ развивается постепенно после переохлаждения, ОРВИ, ангины, иногда начинается с кратковременного повышения температуры тела до 37 – 38 °С. Формируются симметричные диффузные мышечные атрофии, возникают двигательные нарушения в проксимальных отделах конечностей, мышцах плечевого и тазового пояса с обязательным вовлечением мышц дистальных отделов верхних конечностей, рано развиваются фиброз и ретракции аддукторно-флексорно-ротатарной локализации. Болевой синдром не характерен. Мышцы лица не поражаются. Клинически наблюдается сходство с миопатией. При электромиографии (ЭМГ) выявляется миогенно-неврогенный тип поражения. Течение заболевания медленное, неуклонно прогрессирующее, приводящее к тяжелым двигательным нарушениям. Существует несколько вариантов его течения: по типу плечелопаточно-лицевой формы миодистрофии и конечностно-поясничных форм миопатий.
Псевдомиастеническая форма ПМ характеризуется сочетанием полимиозита с миастеническими явлениями. Нередко наблюдаются серьезные нарушения функций мышц лица, страдает мимическая мускулатура, поражаются дыхательные мышцы, развиваются дыхательные кризовые состояния вплоть до необходимости проведения трахеостомии. При ЭМГ выявляются неврогенно-миогенные признаки и утомляемость миастенического типа. Эта форма ПМ отличается тяжелым течением и плохим прогнозом.
Псевдоамиотрофическая форма ПМ характеризуется развитием диффузных мышечных атрофий, тяжелыми двигательными расстройствами, вплоть до невозможности передвигаться. У таких больных часто наблюдаются диспластические признаки – кифосколиоз, вогнутая форма грудной клетки. Отмечаются диффузная гипотония мышц и арефлексия. Иногда наблюдаются кожные высыпания по типу экссудативного диатеза, легкая пастозность лица. При ЭМГ выявляются миогенно-неврогенные признаки поражения. Течение тяжелое, иногда развивается полная обездвижимость.
Миосклеротическая форма ПМ встречается редко и характеризуется бурным развитием миосклероза и контрактур в начале заболевания. У пациентов формируются распространенные ретракции, фиксация конечностей, обездвиженность. В процесс вовлекаются дыхательные мышцы. Наблюдается поражение кожи. При ЭМГ выявляются миогенно-неврогенные изменения.
Миалгическая форма ПМ — встречается наиболее часто (34,9 % случаев). Чаще болеют женщины в возрасте от 15 до 45 лет. Основные проявления – сильные боли в мышцах и боли невралгического характера. Все симптомы ПМ, кроме болевого, представлены незначительно. Кожные покровы не поражаются. Страдает периферическая нервная система. Имеются эндокринные, вегетативные расстройства, арефлексия, полиневритический тип расстройств чувствительности. При ЭМГ выявляется преимущественно неврогенный тип изменений. Течение болезни благоприятное.
Форма с синдромом Мак-Ардля — редкий вариант полимиозита. Характеризуется развитием симптомокомплекса, имеющего сходство с гликогенозом V типа (болезнью Мак-Ардля), связанного с дефицитом мышечной фосфорилазы – фермента, участвующего в процессе гликогенолиза. При этом заболевании развивается ригидность в мышцах при физической нагрузке, сопровождающейся увеличением объема мышц, развитием утомляемости и слабости в сочетании с амиотрофическим синдромом, кожными проявлениями и признаками воспаления. При ЭМГ выявляется миогенно-неврогенный тип изменений.
Осложнения. Наиболее частое и грозное осложнение (занимает первое место среди причин смерти больных ДМ (ПМ)) – аспирация пищи при нарушении глотания и развитие тяжелой аспирационной пневмонии на фоне ограниченной подвижности грудной клетки вследствие поражения межреберных мышц и диафрагмы. Гиповентиляция легких также создает предпосылки к развитию пневмонии вследствие интеркуррентной инфекции. В отдельных случаях тяжелое поражение дыхательных мышц с резким ограничением экскурсии грудной клетки может вести к нарастающей дыхательной недостаточности и асфиксии, что требует применения ИВЛ. Сердечная и особенно почечная недостаточность при ДМ (ПМ) относительно редки. У обездвиженных больных часто возникают язвы, пролежни, которые легко инфицируются; возможны дистрофия, истощение.
Дерматомиозит (ПМ) и беременность. Влияние ДМ (ПМ) на развитие беременности изучено мало. Согласно отдельным наблюдениям и ретроспективным исследованиям, ДМ (ПМ) неблагоприятен как для беременной, так и для плода, поэтому считается, что беременность больным ДМ (ПМ) противопоказана. Так, по данным В. И. Мазурова и И. Б. Беляевой (2005), из 10 беременностей у женщин, больных ДМ (ПМ), 3 закончились спонтанным абортом в период до 20 недель беременности, другие 3 – внутриутробной смертью плода на 21 – 37-й неделе беременности и 4 – преждевременными родами.
Ретроспективное изучение 77 беременностей у 22 % больных ДМ и ПМ до развития заболевания позволило выявить спонтанные аборты лишь у 9 %, перинатальную смерть – в 2,5 % и преждевременные роды – в 3,8 % случаев.
Среди 18 наблюдавшихся женщин у трех ДМ (ПМ) развился в первые 3 мес. беременности и у одной больной – после кесарева сечения. Обострения болезни (в первые 3 мес. беременности – у двух и после аборта – у одной больной) отмечены у других 7 женщин с ДМ (ПМ) в неактивной фазе.
Более обнадеживающие результаты ретроспективного анализа беременности на фоне ДМ и ПМ приводятся Н. Г. Гусевой (2004). Авторы подчеркивают необходимость лечения кортикостероидами в случае обострения ДМ (ПМ), что способствует благоприятному завершению беременности. Наблюдаются отдельные случаи развития острого ПМ во время беременности. При этом терапия кортикостероидами дала быстрый эффект и исход беременности оказался благоприятным как для матери, так и для плода (в том числе с рождением близнецов). По данным Н. Г. Гусевой (2004), лечение беременных, больных ПМ и ДМ, кортикостероидами по показаниям (т. е. при активности процесса), как и прием поддерживающих доз, можно продолжить, так как это практически не влияет на течение беременности. Однако прием нестероидных противовоспалительных препаратов (НПВП) и цитостатиков должен быть прекращен. Больные должны наблюдаться ревматологом и акушером-гинекологом как в течение беременности, так и в послеродовой период (осмотр и проведение необходимых анализов не реже 1 раза в месяц).